Allgemeinheit
Der Begriff Retinitis pigmentosa (RP) identifiziert eine Gruppe genetischer Erkrankungen, die durch eine fortschreitende Netzhautdegeneration gekennzeichnet sind.

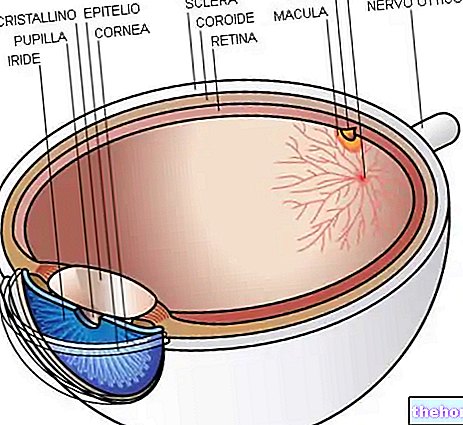
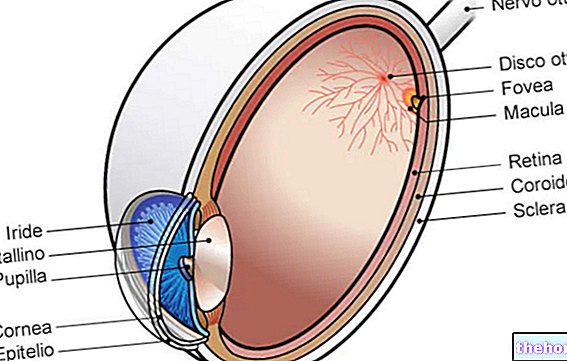
Retinitis pigmentosa ist eine Netzhautdystrophie, die durch den allmählichen Verlust von Photorezeptoren und eine Dysfunktion des Pigmentepithels gekennzeichnet ist, was bedeutet, dass die Netzhaut ihre Fähigkeit, visuelle Informationen über den Sehnerv an das Gehirn zu übermitteln, zunehmend verringert.
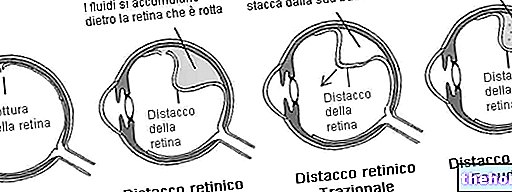
Der pathologische Prozess beginnt mit Veränderungen des retinalen Pigmentepithels. Mit fortschreitender Retinitis pigmentosa kommt es zu einer Verdünnung der die Netzhaut versorgenden Blutgefäße, die atrophieren. Bei der Untersuchung des Fundus sind die charakteristischen Ablagerungen visuell erkennbar. daher der Name von der Krankheit). Atrophische Veränderungen und Schäden können auch den Sehnerv betreffen und allmählich sterben die lichtempfindlichen Zellen der Netzhaut ab.
Patienten mit Retinitis pigmentosa haben vor allem in schlecht beleuchteten Umgebungen zunächst Sehstörungen und klagen über eine Einengung des peripheren Gesichtsfeldes. Das zentrale Sehvermögen bleibt bis in die späteren Stadien der Erkrankung verschont, und das Endergebnis kann dramatisch variieren: Viele Menschen mit Retinitis pigmentosa behalten ihr ganzes Leben lang eine eingeschränkte Sehkraft, während andere das Sehvermögen vollständig verlieren.
Retinitis pigmentosa ist eine Erbkrankheit, die hauptsächlich durch genetische Veränderungen verursacht wird, die von einem oder beiden Elternteilen weitergegeben werden. Die Art des genetischen Defekts bestimmt, welche Netzhautzellen am stärksten an der Erkrankung beteiligt sind und ermöglicht es, aus klinischer Sicht die verschiedenen Erkrankungen zu unterscheiden. Bis heute wurden mehr als 50 verschiedene genetische Defekte identifiziert, die mit der Retinitis pigmentosa in Verbindung gebracht werden. Anomalien können durch eines von drei Vererbungsmustern von den Eltern an die Nachkommen weitergegeben werden: autosomal-rezessiv, autosomal-dominant oder heterosomal-rezessiv (X-chromosomal oder X-chromosomal).
Symptome
Für weitere Informationen: Retinitis pigmentosa Symptome
Die Retinitis pigmentosa tritt meist bei Jugendlichen und jungen Erwachsenen auf. Die Symptome treten oft im Alter zwischen 10 und 30 Jahren auf, aber die Diagnose kann in der frühen Kindheit oder viel später im Leben gestellt werden.
Frühe Symptome einer Retinitis pigmentosa können sein:
- Schwierigkeiten beim Sehen bei Nacht (Nachtblindheit) oder bei schlechten Lichtverhältnissen
- Langsame Anpassung vom Sehen im Dunkeln zum Hellen und umgekehrt;
- Verengung des Gesichtsfeldes und Verlust des peripheren Sehens;
- Licht- und Blendempfindlichkeit.
Einige Symptome hängen von der Art der beteiligten Photorezeptoren ab. Die Stäbchen sind für das Schwarz-Weiß-Sehen verantwortlich, während die Zapfen Ihnen die Unterscheidung von Farben ermöglichen.
Bei der Retinitis pigmentosa sind in den meisten Fällen zuerst die Stäbchen betroffen. Bei den sich schnell entwickelnden Formen können jedoch auch Zapfen frühzeitig betroffen sein.
Die Stäbchen sind in den äußeren Teilen der Netzhaut konzentriert und werden durch schwaches Licht aktiviert, so dass ihre Degeneration die periphere und Nachtsicht beeinträchtigt. Wenn Zapfen beteiligt sind, kann es zu einem Verlust der Farbwahrnehmung und des zentralen Sehens kommen.
Die Dominanz der beteiligten Photorezeptoren wird durch den jeweiligen Defekt in der genetischen Ausstattung des Patienten bestimmt.
Das erste Symptom der Retinitis pigmentosa ist oft Nachtblindheit (oder Nokthalopie). Manche Menschen stellen fest, dass sie immer mehr Zeit brauchen, um sich an Lichtunterschiede anzupassen, wenn sie von einem gut beleuchteten Bereich zu einem dunkleren wechseln. Eine typische Form des Sehverlusts führt zu einer Verengung des peripheren Sehens (Tunnel- oder Fernrohrblick); Dieses Muster wird als Ringskotom bezeichnet. Manchmal kann dieses Phänomen im Frühstadium fehlen, aber es wird bemerkt, wenn die Person oft über Gegenstände stolpert oder in einen Verkehrsunfall verwickelt ist.Wenn der Sehverlust den zentralen Bereich der Netzhaut betrifft (auch Makuladystrophie genannt) Patienten Schwierigkeiten beim Lesen und Detailarbeit haben, die die Konzentration auf einen einzigen Gegenstand erfordert, wie z. B. das Einfädeln eines Fadens durch ein Nadelöhr Viele Patienten berichten von Lichtblitzen (Photopsie), die oft als kleine, flackernde und funkelnde Lichter beschrieben werden.
Die Geschwindigkeit des Krankheitsverlaufs und der Grad des Sehverlusts sind von Person zu Person unterschiedlich. Einige Extremfälle können sich innerhalb von zwei Jahrzehnten schnell entwickeln, andere ein langsamer Verlauf, der nie zur vollständigen Erblindung führt. Ein früher Beginn tritt bei schwereren Formen der Retinitis pigmentosa auf, während Patienten mit leichteren Erkrankungen (z. B. autosomal-dominant) die Krankheit im fünften oder sechsten Lebensjahrzehnt entwickeln können.In Familien mit X-chromosomaler Retinitis pigmentosa sind Männer häufiger betroffen als Frauen und schwerer; Frauen hingegen übertragen das genetische Merkmal (sie tragen das veränderte Gen auf dem X-Chromosom) und zeigen seltener Symptome der Erkrankung.
Komplikationen
Die Retinitis pigmentosa wird weiter fortschreiten, wenn auch langsam. Eine vollständige Erblindung ist jedoch selten, aber es kann zu einer signifikanten Einschränkung des peripheren und zentralen Sehens kommen.
Patienten mit Retinitis pigmentosa entwickeln oft schon in jungen Jahren eine Netzhautschwellung (Makulaödem) oder einen grauen Star. Diese Komplikationen können behandelt werden, wenn sie das Sehvermögen beeinträchtigen.
Verwandte Krankheiten
Üblicherweise hat ein Patient mit Retinitis pigmentosa keine weiteren Erkrankungen und in diesem Fall spricht man von "nicht-syndromaler" oder einfacher Retinitis pigmentosa. Einige Syndrome teilen jedoch einige klinische Symptome mit dieser Augenerkrankung; am häufigsten ist das Usher-Syndrom, das etwa 10-30 % aller Patienten mit Retinitis pigmentosa betrifft und mit gleichzeitigem angeborenem oder fortschreitendem Hörverlust einhergeht. Bei der angeborenen Amaurose Leber können Kinder jedoch innerhalb der ersten sechs Lebensmonate erblinden oder fast erblinden.Weitere Erkrankungen, die mit der Retinitis pigmentosa in Zusammenhang stehen, sind das Bardet-Biedl-Syndrom und der Morbus Refsum.
Ursachen
Die Krankheit kann durch eine Reihe von genetischen Defekten verursacht werden: Tatsächlich gibt es mehrere Gene, die, wenn sie von der Veränderung betroffen sind, den Phänotyp der Retinitis pigmentosa verursachen können.Diese normalerweise kodieren Proteine, die an der Transduktionskaskade beteiligt sind, die das Sehen ermöglicht und die Zelltranskription beeinflusst (die fehlerhafte Nachrichten an Netzhautzellen senden) oder für Elemente, die die Struktur von Photorezeptoren ausmachen. Vererbte Genmutationen sind in Zellen vom Moment der Empfängnis an vorhanden; häufige Anomalien umfassen die der RP1-Gene (bei Retinitis pigmentosa-1, autosomal dominant) , RHO (RP4, autosomal dominant) und RDS (RP7, autosomal dominant) Nicht-erbliche Ursachen der Retinitis pigmentosa sind selten, aber die Möglichkeit, einen Einzelfall (Spontanmutation) zu finden, bei dem sie nicht vorhanden ist eine Familienanamnese von die Krankheit.