Diese Krankheit verdankt ihren Namen dem amerikanischen Endokrinologen, der sie entdeckte: Frederic Crosby Bartter Die jährliche Inzidenz wird auf 1 / 830.000 geschätzt.
Es gibt mehrere Varianten des Bartter-Syndroms, deren Vererbung, obwohl noch autosomal, je nach Fall von rezessiv bis dominant variieren kann.
Wird das Bartter-Syndrom nicht rechtzeitig diagnostiziert und behandelt, kann dies die Entwicklung, das Wachstum und die Lebensqualität des Patienten stark beeinträchtigen. Außerdem wird in besonders schweren Fällen die Lebenserwartung erheblich verkürzt.
Bitte beachten Sie
Das Bartter-Syndrom sollte NICHT mit dem Schwartz-Bartter-Syndrom verwechselt werden, einer Krankheit, die durch eine „gestörte Sekretion des antiduiretischen Hormons (ADH) gekennzeichnet ist, auch bekannt als das Syndrom der inadäquaten ADH-Sekretion (SIADH).
.die auf der Ebene der Henle-Schleife auftritt, ist auf eine "Änderung der Synthese einiger Kanal- / Transporterrezeptoren (insbesondere Proteine, die Ionen unterschiedlicher Natur transportieren) in diesem" Bereich der Niere zurückzuführen. Dieses Phänomen wird verursacht durch eine Reihe von genetischen Mutationen, die die Gene betreffen, die für die oben genannten bestimmten Proteine kodieren.
Die verschiedenen Varianten des Bartter-Syndroms werden nach dem betroffenen Gen unterschieden. Nähere Informationen hierzu finden Sie im folgenden Kapitel.
.
Die folgende Tabelle zeigt daher die verschiedenen Varianten des Syndroms, die beteiligten mutierten Gene, die Proteine (Kanalrezeptoren / Transporter), für die sie kodieren, und die klinische Präsentation der jeweiligen Variante.
Variante
Mutiertes Gen
Kanal / Transporter beteiligt
Klinische Präsentation
Bartter-Syndrom Typ I
Gen SLC12A1
NKCC2 (Natrium-Kalium-Chlor-Cotransporter oder Na + / K + / 2Cl-)
Pränatales (oder infantiles) Bartter-Syndrom
Bartter-Syndrom Typ II
Gen KCNJ1
ROMK (Kaliumkanal des äußeren Nierenmarks)
Pränatales (oder infantiles) Bartter-Syndrom
Bartter-Syndrom Typ III
Gen CLNKb
CLCNKb (Chlorkanal vom Typ Kb)
Klassisches Bartter-Syndrom
Bartter-Syndrom Typ IV oder IV A
Gen BSND
Barttina (Beta-Untereinheit von Chlorkanälen vom Ka- und Kb-Typ)
Pränatales (oder infantiles) Bartter-Syndrom und Schallempfindungsschwerhörigkeit
Bartter-Syndrom Typ IV B
CLCNKa- und CLCNKb-Gene
CLCNKa (Chlorkanal Typ Ka) und CLCNKb
Pränatales (oder infantiles) Bartter-Syndrom und sensorineurale Taubheit
Bartter-Syndrom Typ V
CASR-Gen
CaSR (calciumsensitiver Rezeptor)
Bartter-Syndrom mit Hypokalzämie
Wie aus der Tabelle ersichtlich, können trotz des Vorhandenseins von fünf genetischen Varianten nicht so viele klinische Formen unterschieden werden; tatsächlich werden nur vier unterschieden: pränatales oder infantiles Bartter-Syndrom (Typ I und II), klassisches Bartter-Syndrom (Typ III), pränatales oder infantiles Bartter-Syndrom mit sensorineuraler Taubheit (Typ IV A und IV B; einige Quellen jedoch sie gruppieren diese Varianten mit Typ I und II) und schließlich das Bartter-Syndrom mit Hypokalzämie (Typ V).
Wussten Sie, dass ...
Angesichts der Existenz einer Variante IV (oder IV A) und einer Variante IV B des Bartter-Syndroms betrachten einige Quellen insgesamt sechs Varianten des Bartter-Syndroms, andere Quellen hingegen betrachten die Variante IV B als Subtyp der Variante IV und , aus diesem Grund die Existenz von nur fünf genetischen Varianten des Bartter-Syndroms in Betracht ziehen.
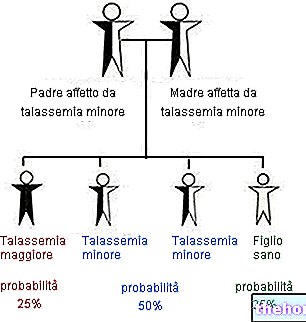
Varianten von Typ I, II, III, IV und IV B sind autosomal-rezessiv übertragene Krankheiten, was bedeutet, dass das Individuum zur Manifestation des Syndroms beide mutierten Allele besitzen muss, die von den Eltern geerbt werden, die daher gesunde Träger sind des Syndroms hingegen ist eine autosomal-dominant vererbte Krankheit, d.h. zur Manifestation der Symptome reicht es aus, wenn der Patient ein einziges mutiertes Allel besitzt, das also auch nur von einem (auch krank) vererbt werden kann ) der beiden Eltern.
Bartters Pseudosyndrom
Das Bartter-Pseudo-Syndrom ist ein Zustand, der durch ähnliche Symptome wie beim Bartter-Syndrom gekennzeichnet ist, deren Ursache jedoch im Missbrauch von Diuretika wie Furosemid zu suchen ist.
Gitelman-Syndrom
Dieses Syndrom wird durch eine lokalisierte Mutation im SLC12A3-Gen verursacht, das für den Natrium-Chlor-Cotransporter (NCC) kodiert. Durch diese autosomal-rezessiv vererbte Mutation kommt es beim Patienten zu einer Beeinträchtigung der Rückresorption von Natrium, Chlor und Kalium auf der Ebene des distalen Tubulusfaltens, im Gegensatz zum Bartter-Syndrom, bei dem die Beeinträchtigung der Resorption im „Allerdings“ lokalisiert ist , kann das Gitelman-Syndrom ähnliche Symptome wie das Bartter-Syndrom hervorrufen, weshalb es in der klinischen Praxis manchmal schwierig sein kann, die beiden Krankheiten zu unterscheiden.
, Hypochlorämie und metabolische Alkalose, die mit Hyperreninämie (Renin im Blut) und Hyperaldosteronismus einhergehen können. Natürlich können alle diese Zustände wiederum eine Reihe von Symptomen hervorrufen, die die Lebensqualität des Patienten beeinträchtigen können (z. B. Übelkeit, Erbrechen, Schwindel, Schwäche, Kopfschmerzen, Hypotonie usw.).
Zusätzlich zu dem bisher Gesagten kann jede Variante zu spezifischen Manifestationen und Symptomen führen, die eng mit dem mutierten Gen und der daraus folgenden Beteiligung des Kanals oder Cotransporters, für den dieses Gen kodiert, verbunden sind. Daher werden im Folgenden die typischen Symptome und Manifestationen, die mit jeder der fünf verschiedenen Formen des Bartter-Syndroms verbunden sind, kurz beschrieben.
Bartter-Syndrom Typ I
Beim Bartter-Syndrom Typ I betreffen die Mutationen das Gen, das für den Natrium-Kalium-Chlor-Cotransporter auf der Henle-Schleife kodiert.Durch die beeinträchtigte Rückresorption kommt es zu einer Hypovolämie durch Salzverlust. Da gleichzeitig auch die Resorption von Calcium mit der Aktivität des oben genannten Cotransporters verbunden ist, erleben wir den Beginn einer Hyperkalziurie, die zur Entstehung einer Nephrokalzinose führen kann. Es ist auch möglich, Hypermagnesurie zu erleben. Polyhydramnion als Folge einer fetalen Polyurie kann sich in der pränatalen Phase entwickeln.
Bartter-Syndrom Typ II
Das Bartter-Syndrom Typ II wird durch eine Mutation im Gen verursacht, das für den Kaliumkanal des Nebennierenmarks kodiert. Manifestationen und Symptome ähneln denen der Variante I und auch hier kann man als Folge einer fetalen Polyurie auf Polyhydramnion stoßen. In einem frühen Stadium kann das Neugeborene jedoch eine vorübergehende hyperkaliämische metabolische Azidose erleiden. Dieser Zustand entwickelt sich dann zum charakteristischen klinischen Bild des Bartter-Syndroms.
Bartter-Syndrom Typ III
Die auch als klassisches Bartter-Syndrom bekannte Variante III der Krankheit wird durch Mutationen im Gen, das für den Chlorkanal vom Kb-Typ kodiert, verursacht.Da die Chlorkanäle vom Ka-Typ in dieser Form erhalten bleiben, sind die Symptome tendenziell leichter, obwohl sie noch vorhanden sind. Eine Nephrokalzinose liegt im Allgemeinen nicht vor.
Bartter-Syndrom Typ IV und IV B
Bei beiden Typen der Variante IV sind Gene beteiligt, die an der korrekten Synthese der Ka- und Kb-Chlorkanäle beteiligt sind.Da beide Kanäle beeinträchtigt sind, sind die Symptome tendenziell schwerwiegender als bei Variante III des Syndroms. Säuglinge können zunächst ein Krankheitsbild zeigen, das einen Hypoaldosteronismus nachahmt, sich dann aber zu einer hypokaliämischen metabolischen Alkalose entwickelt, wenn der Körper versucht, die fehlende Aktivität der oben genannten Kalziumkanäle auszugleichen.Kennzeichnend für die Varianten IV und IV B des Bartter-Syndroms ist das Auftreten von sensorineuralen Taubheit.
Bartter-Syndrom Typ V
Die Variante V des Bartter-Syndroms wird durch eine Mutation verursacht, die das Gen betrifft, das für den kalziumsensitiven Rezeptor kodiert, der an der Hemmung der Rückresorption von Wasser und verschiedenen Ionen wie Kalzium, Kalium und Natrium beteiligt ist daraus resultierende Hyperkalziurie in Verbindung mit den charakteristischen Symptomen des Bartter-Syndroms.
Wussten Sie, dass ...
Die Varianten I, II, IV und IV B des Bartter-Syndroms – sowie unter der Bezeichnung pränatales Bartter-Syndrom – werden manchmal auch als Hypeprostaglandin-E2-Syndrom bezeichnet, da sie durch einen Anstieg der Plasmaspiegel dieses Prostaglandins gekennzeichnet sind.
- zum Nachweis des Vorhandenseins und der Konzentration von Elektrolyten (Natrium, Kalium, Chlorid, Magnesium, Bicarbonat, Calcium) und bestimmten Substanzen (Renin und Aldosteron) im Plasma und / oder Urin.Die definitive Diagnose ist jedoch nur durch die Durchführung spezifischer genetischer Tests möglich.
Die Differenzialdiagnose muss hingegen gegen das Bartter-Pseudo-Syndrom, das Gitelman-Syndrom, die Mukoviszidose und die Zöliakie gestellt werden.
In Fällen, in denen ein gewisses Risiko besteht (z. B. Eltern mit gesunden und/oder kranken Trägern), dass das Neugeborene die Krankheit manifestiert, ist auch eine pränatale Diagnostik möglich.
von:
- Ergänzungen mit Mineralsalzen (insbesondere, aber nicht ausschließlich, Kalium) zum Ausgleich der fehlenden Rückresorption;
- Nichtsteroidale Antirheumatika (NSAR) wie zB Indomethacin Diese Medikamente werden mit dem Ziel verabreicht, zu hohe Prostaglandin-E2-Spiegel zu senken;
- Kaliumsparende Diuretika (zur Verringerung der Kaliumausscheidung im Urin).
In den schwerwiegendsten Fällen und / oder bei Stresszuständen (Beginn anderer Krankheiten, chirurgische Eingriffe usw.) kann die Auffüllung von Kalium und anderen Mineralsalzen intravenös erfolgen, natürlich muss eine ähnliche Operation von der Gesundheit durchgeführt werden Personal spezialisiert.