Was ist Phenylketonurie?
Dort Phenylketonurie (P.K.U.) Es handelt sich um eine autosomal-rezessiv vererbte Stoffwechselerkrankung, die 1 von 10.000 Personen betrifft und häufiger bei Homozygoten als bei Heterozygoten aufzutreten scheint.
Die zur Gruppe der Hyperphenylalaninämie gehörende Phenylketonurie beeinträchtigt den Stoffwechsel von Phenylalanin und insbesondere dessen Umwandlung in Tyrosin; Phenylketonurie wird durch die erhöhten Urinspiegel von Phenylalanin und einigen Derivaten (Phenylpyruvat, Phenylacetat, Phenylactat und Phenylacetylglutamin) erkannt.
Die schwerwiegendste Komplikation der Phenylketonurie ist die geistige Verzögerung.
Phenylalanin, Tyrosin und Derivate
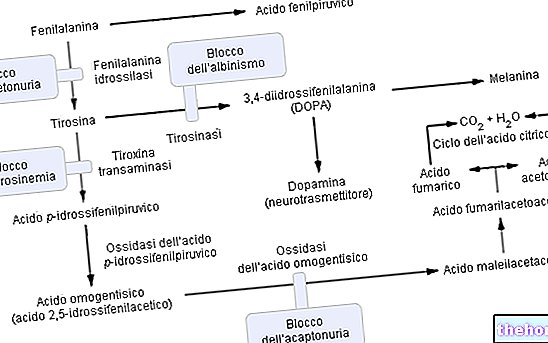
Phenylalanin ist eine essentielle Aminosäure und bildet den Großteil der Nahrungsproteine; es kann durch das Enzym umgewandelt werden Phenylalanin-Hydroxylase in Tyrosin (durch Hinzufügen einer Hydroxylgruppe -OH). Tyrosin wiederum ist eine Vorläuferaminosäure für die Synthese von:
- L-DOPA (Dopaminsynthese-Zwischenprodukt)
- Adrenalin
- Noradrenalin (alle Neurotransmitter).

Mechanismus der Phenylketonurie (P.K.U.)
Wie erwartet, ist bei Phenylketonurie aufgrund einer oder mehrerer (insgesamt 6) Chromosomenmutationen die Expression (daher die Stoffwechselaktivität) der Phenylalanin-Hydroxylase praktisch null. Diese Veränderungen können unterschiedlicher Art sein (von "missense"-Änderungen über "spleißende" Defekte oder sogar "partielle Deletionen"), aber was zählt, ist, dass aufgrund dieser enzymatischen Ineffizienz die Phenylalaninspiegel im Blut (die normalerweise 1 mg / 100 ml betragen) bei DOMINANT Phenylketonurie Sie erreichen problemlos sogar 50-mal höhere Mengen.
Funktionsweise des Enzyms Phenylalanin-Hydroxylase: Um Tyrosin (+ Dihydrobiopterin) zu produzieren, benötigt Phenylalanin-Hydroxylase: Phenylalanin, Sauerstoff und Tetrahydrobiopterin (ein reduziertes Pteridin, das als Coofaktor wirkt); die Reaktion ist auch reversibel und das Dihydrobiopterin kann wieder umgewandelt werden (dank des Enzyms Dihydropterinreduktase) in Tetrahydrobiopterin.
Komplikationen
Die Phenylketonurie kann je nach Schwere der pathologischen Manifestation und der Rechtzeitigkeit der Diagnose zu mehr oder weniger schweren Komplikationen führen; Als erbliche Pathologie wird die Phenylketonurie unterschieden in:
- Dominant, daher gekennzeichnet durch VOLLSTÄNDIGE Inaktivität des Phenylalanin-Hydroxylase-Enzyms
- Rezessiv, bei dem nur 30% des gesamten enzymatischen Erbes aktiv sind.
Die Komplikationen der Phenylketonurie sind direkt proportional auf die metabolische Akkumulation von Phenylalanin, seinen Derivaten und die verminderte Tyrosinsynthese zurückzuführen.In der Pathologie wird das überschüssige Phenylalanin relativ effektiv von der Niere gefiltert, die es nur teilweise resorbiert und mit dem Urin ausscheidet ; jedoch bestimmt die Persistenz der Hyper-Phenylalaninämie-Spiegel eine metabolische Reaktion der molekularen Umwandlung in Phenylbrenztraubensäure und/oder andere leichter entwässernde Derivate (Phenylpyruvat, Phenylacetat, Phenylactat).
Was die Phenylketonurie erschwert, ist die Toxizität von Phenylalanin, Phenylbrenztraubensäure und ihren Derivaten gegenüber dem Zentralnervensystem (ZNS), deren übermäßige Präsenz in der Gehirnentwicklung unaufhaltsam eine Form der geistigen Behinderung bestimmt.
Achtung. Die Plasmakonzentrationen der anderen Aminosäuren sind leicht reduziert, wahrscheinlich aufgrund von Rückkopplungen zur intestinalen Resorption oder renalen tubulären Reabsorption.
Hirnschäden als schwerwiegende Komplikation der Phenylketonurie werden durch die Subtraktion anderer essentieller Aminosäuren bei der Proteosynthese verursacht, insbesondere bei der Bildung von Polyribosomen, Myelin, Noradrenalin und Serotonin. Phenylketonurie – nicht unmittelbar nach der Geburt, aber nach einigen Jahren sichtbar – erfordert, wenn sie nicht behandelt wird, einen Krankenhausaufenthalt des Kindes und ist vollständig irreversibel.
Eine fortgeschrittene Phenylketonurie kann auch mit bloßem Auge deutlich sichtbar sein; die hohen Konzentrationen von Phenylalanin, die das Enzym hemmen Tyrosinase, die Melaninsynthese erheblich beeinträchtigen, indem sie die Pigmentierung von Haut und Haaren reduzieren; außerdem verleiht die Anreicherung von Phenylacetat in Haar und Haut den Phenylketonurika einen starken und unangenehmen "Mausgeruch".