Wirkstoffe: Anastrozol
Arimidex 1 mg Filmtabletten
Warum wird Arimidex verwendet? Wofür ist das?
Arimidex enthält eine Substanz namens Anastrozol, die zu einer Gruppe von Arzneimitteln gehört, die als „Aromatasehemmer“ bezeichnet werden. Arimidex wird zur Behandlung von Brustkrebs bei postmenopausalen Frauen angewendet.
Arimidex wirkt, indem es die vom Körper produzierte Menge an Hormonen, den sogenannten Östrogenen, reduziert, indem es eine natürliche Substanz (Enzym) in Ihrem Körper, die sogenannte „Aromatase“, blockiert.
Kontraindikationen Wenn Arimidex nicht verwendet werden sollte
Arimidex darf nicht eingenommen werden:
- wenn Sie allergisch gegen Anastrozol oder einen der sonstigen Bestandteile dieses Arzneimittels sind
- wenn Sie schwanger sind oder stillen (siehe Abschnitt „Schwangerschaft und Stillzeit“).
Nehmen Sie Arimidex nicht ein, wenn einer der oben genannten Punkte auf Sie zutrifft. Fragen Sie im Zweifelsfall Ihren Arzt oder Apotheker, bevor Sie Arimidex einnehmen.
Vorsichtsmaßnahmen für die Anwendung Was sollten Sie vor der Einnahme von Arimidex beachten?
Bitte sprechen Sie mit Ihrem Arzt, Apotheker oder dem medizinischen Fachpersonal, bevor Sie Arimidex einnehmen:
- wenn Sie noch menstruieren und sich noch nicht in den Wechseljahren befinden.
- wenn Sie ein Arzneimittel einnehmen, das Tamoxifen oder Östrogen enthält (siehe Abschnitt „Einnahme von ARIMIDEX mit anderen Arzneimitteln“).
- wenn Sie eine Erkrankung haben oder jemals hatten, die die Festigkeit Ihrer Knochen beeinträchtigt (Osteoporose).
- wenn Sie Leber- oder Nierenprobleme haben.
Wenn Sie sich nicht sicher sind, ob einer der oben genannten Punkte auf Sie zutrifft, fragen Sie vor der Einnahme von Arimidex Ihren Arzt oder Apotheker.
Wenn Sie ins Krankenhaus eingeliefert werden, teilen Sie dem medizinischen Personal mit, dass Sie Arimidex einnehmen.
Wechselwirkungen Welche Medikamente oder Lebensmittel können die Wirkung von Arimidex® beeinflussen?
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen oder kürzlich andere Arzneimittel eingenommen haben.
Dazu gehören Medikamente, die Sie ohne Rezept kaufen können, und pflanzliche Heilmittel. Dies liegt daran, dass Arimidex die Wirkung einiger Arzneimittel beeinflussen kann und einige Arzneimittel eine Wirkung auf Arimidex haben können.
Nehmen Sie Arimidex nicht ein, wenn Sie bereits eines der folgenden Arzneimittel einnehmen:
- Einige Arzneimittel zur Behandlung von Brustkrebs (selektive Östrogenrezeptormodulatoren), zum Beispiel Arzneimittel, die Tamoxifen enthalten. Dies liegt daran, dass diese Arzneimittel die ordnungsgemäße Wirkung von Arimidex beeinträchtigen können.
- Östrogenhaltige Arzneimittel, zum Beispiel Hormonersatztherapie (HRT).
Wenn einer dieser Punkte auf Sie zutrifft, fragen Sie Ihren Arzt oder Apotheker um Rat. Informieren Sie Ihren Arzt oder Apotheker, wenn Sie Folgendes einnehmen:
- Ein als "LHRH-Analogon" bekanntes Arzneimittel. Dazu gehören Gonadorelin, Buserelin, Goserelin, Leuprorelin und Triptorelin. Diese Arzneimittel werden zur Behandlung von Brustkrebs, bestimmten weiblichen (gynäkologischen) Erkrankungen und Unfruchtbarkeit angewendet.
Warnungen Es ist wichtig zu wissen, dass:
Schwangerschaft und Stillzeit
Nehmen Sie Arimidex nicht ein, wenn Sie schwanger sind oder stillen. Beenden Sie Arimidex, wenn Sie schwanger werden, und sprechen Sie mit Ihrem Arzt.
Fragen Sie vor der Einnahme von Arzneimitteln Ihren Arzt oder Apotheker um Rat.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Es ist unwahrscheinlich, dass Arimidex Ihre Verkehrstüchtigkeit oder Ihre Fähigkeit zum Bedienen von Werkzeugen oder Maschinen beeinträchtigt. Einige Personen können sich jedoch während der Einnahme von Arimidex gelegentlich schwach oder schläfrig fühlen.Fragen Sie in diesem Fall Ihren Arzt oder Apotheker um Rat.
Arimidex enthält Lactose
Arimidex enthält Lactose, eine Zuckerart. Bitte nehmen Sie dieses Arzneimittel erst nach Rücksprache mit Ihrem Arzt ein, wenn Ihnen bekannt ist, dass Sie unter einer Zuckerunverträglichkeit leiden.
Dosis, Methode und Zeitpunkt der Verabreichung Wie ist Arimidex anzuwenden: Dosierung
Nehmen Sie Arimidex immer genau nach Absprache mit Ihrem Arzt oder Apotheker ein. Fragen Sie im Zweifelsfall Ihren Arzt oder Apotheker.
- Die empfohlene Dosis beträgt einmal täglich eine Tablette.
- Versuchen Sie, die Tablette jeden Tag zur gleichen Zeit einzunehmen.
- Schlucken Sie die Tablette im Ganzen mit einem Schluck Wasser.
- Es spielt keine Rolle, ob Sie Arimidex vor, mit oder nach den Mahlzeiten einnehmen.
Nehmen Sie Arimidex so lange ein, wie Ihr Arzt oder Apotheker es Ihnen sagt. Es handelt sich um eine Langzeitbehandlung, die möglicherweise über mehrere Jahre eingenommen werden muss. Fragen Sie im Zweifelsfall Ihren Arzt oder Apotheker.
Anwendung bei Kindern und Jugendlichen
Arimidex darf Kindern und Jugendlichen nicht verabreicht werden.
Überdosierung Was ist zu tun, wenn Sie zu viel Arimidex eingenommen haben?
Wenn Sie eine größere Menge von Arimidex eingenommen haben, als Sie sollten
Wenn Sie eine größere Menge von Arimidex eingenommen haben, als Sie sollten, informieren Sie sofort Ihren Arzt.
Wenn Sie die Einnahme von Arimidex vergessen haben
Wenn Sie die Einnahme einer Dosis vergessen haben, nehmen Sie einfach die nächste Dosis wie gewohnt ein. Nehmen Sie nicht die doppelte Dosis (zwei Dosen gleichzeitig) ein, wenn Sie die vorherige Einnahme vergessen haben.
Wenn Sie die Einnahme von Arimidex® abbrechen
Brechen Sie die Einnahme der Tabletten nicht ab, es sei denn, Ihr Arzt sagt es Ihnen.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt, Apotheker oder das medizinische Fachpersonal.
Nebenwirkungen Was sind die Nebenwirkungen von Arimidex
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Sehr häufige Nebenwirkungen (betreffen mehr als 1 von 10 Behandelten)
- Kopfschmerzen.
- Hitzewallungen.
- Übelkeit (Übelkeit).
- Ausschlag.
- Gelenkschmerzen oder Steifheit.
- Gelenkentzündung (Arthritis).
- Schwächegefühl.
- Verlust der Knochendichte (Osteoporose).
Häufige Nebenwirkungen (betrifft 1 bis 10 Behandelte von 100)
- Appetitverlust.
- Erhöhte oder erhöhte Blutwerte einer Fettsubstanz namens Cholesterin, die mit einem Bluttest überprüft werden können.
- Schläfrig fühlen.
- Karpaltunnelsyndrom (Kribbeln, Schmerzen, Kältegefühl in Teilen der Hand).
- Kribbeln, Kribbeln oder Taubheitsgefühl der Haut, Geschmacksverlust/-mangel.
- Durchfall.
- Übelkeit (Erbrechen).
- Veränderungen der Blutwerte im Zusammenhang mit der Leberfunktion.
- Ausdünnung der Haare (Haarausfall).
- Allergische (Überempfindlichkeits-) Reaktionen einschließlich des Gesichts, der Lippen und der Zunge.
- Knochenschmerzen.
- Vaginale Trockenheit.
- Vaginale Blutungen (normalerweise in den ersten Behandlungswochen – wenn die Blutung anhält, sprechen Sie mit Ihrem Arzt).
- Muskelschmerzen.
Gelegentliche Nebenwirkungen (betrifft 1 bis 10 Behandelte von 1.000)
- Veränderungen bei bestimmten Bluttests, die zeigen, dass Ihre Leber funktioniert (Gamma-GT und Bilirubin).
- Leberentzündung (Hepatitis).
- Urtikaria.
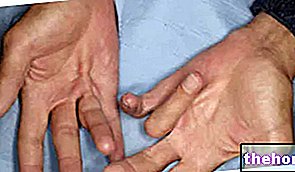
- Schnappfinger (Zustand, in dem einer Ihrer Finger oder Daumen eine gekrümmte Position einnimmt).
- Erhöhen Sie die Menge an Kalzium in Ihrem Blut. Wenn Sie sich krank, erbrechen und durstig fühlen, informieren Sie Ihren Arzt oder Apotheker oder das medizinische Fachpersonal, da möglicherweise Blutuntersuchungen durchgeführt werden müssen.
Seltene Nebenwirkungen (betrifft 1 bis 10 Behandelte von 10.000)
- Ungewöhnliche Entzündung der Haut, die rote Flecken oder Blasen umfassen kann.
- Hauterythem durch Überempfindlichkeit (dies kann durch eine allergische oder anaphylaktische Reaktion verursacht werden).
- Entzündung der Kapillaren, die eine rote oder violette Verfärbung der Haut verursacht. Sehr selten können Gelenk-, Magen- und Nierenschmerzen auftreten; es ist als "Henoch Schönlein purpura" bekannt.
Sehr seltene Nebenwirkungen (betrifft weniger als 1 von 10.000 Behandelten)
- Extrem schwere Hautreaktion mit dem Auftreten von Geschwüren oder Blasen auf der Haut. Es ist als "Stevens-Johnson-Syndrom" bekannt.
- Allergische (Überempfindlichkeits-) Reaktion mit Schwellung des Rachens, die Schluck- oder Atembeschwerden verursachen kann. Bekannt als "Angioödem". Rufen Sie in diesem Fall sofort einen Krankenwagen oder suchen Sie einen Arzt auf – möglicherweise ist eine dringende medizinische Behandlung erforderlich.
Auswirkungen auf die Knochen
Arimidex reduziert die Menge an Hormonen, die Östrogen genannt werden, im Körper. Dies kann den Mineralstoffgehalt der Knochen verringern; daher können die Knochen weniger stark und anfälliger für Frakturen sein. Ihr Arzt wird diese Risiken gemäß den Behandlungsrichtlinien überwachen Gesundheit bei postmenopausalen Frauen. Sprechen Sie mit Ihrem Arzt über diese Risiken und Behandlungsmöglichkeiten.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker, wenn Sie Nebenwirkungen bemerken, die nicht in dieser Packungsbeilage aufgeführt sind.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker, einschließlich aller möglichen Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind. Sie können Nebenwirkungen auch direkt über das nationale Meldesystem auf der Website der italienischen Arzneimittelbehörde melden: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
Ablauf und Aufbewahrung
Nicht über 30°C lagern
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Bewahren Sie die Tabletten an einem sicheren Ort auf, an dem Kinder sie nicht sehen oder erreichen können. Tabletten können für sie gefährlich sein.
Verwenden Sie dieses Arzneimittel nicht nach dem auf der Packung und der Blisterpackung angegebenen Verfallsdatum Das Verfallsdatum bezieht sich auf den letzten Tag des Monats.
Bewahren Sie die Tabletten in der Originalverpackung auf.
Werfen Sie Arzneimittel nicht in das Abwasser oder den Hausmüll. Fragen Sie Ihren Apotheker, wie Sie Arzneimittel, die Sie nicht mehr verwenden, entsorgen. Dies trägt zum Schutz der Umwelt bei.
Was Arimidex enthält
- Der Wirkstoff ist Anastrozol. Jede Filmtablette enthält 1 mg Anastrozol.
- Die sonstigen Bestandteile sind: Lactose-Monohydrat, Povidon, Natriumstärkeglycolat, Magnesiumstearat, Hypromellose, Macrogol 300 und Titandioxid.
Wie Arimidex aussieht und Inhalt der Packung
Die Filmtabletten sind weiß, rund, bikonvex, ca. 6,1 mm groß und mit „A“ auf der einen Seite und „Adx1“ auf der anderen Seite gekennzeichnet.
Arimidex ist in Blisterpackungen mit 28 Tabletten erhältlich.
Quelle Packungsbeilage: AIFA (Italienische Arzneimittelbehörde). Im Januar 2016 veröffentlichter Inhalt. Die vorliegenden Informationen können nicht aktuell sein.
Um Zugriff auf die aktuellste Version zu erhalten, ist es ratsam, auf die Website der AIFA (Italienische Arzneimittelbehörde) zuzugreifen. Haftungsausschluss und nützliche Informationen.
01.0 BEZEICHNUNG DES ARZNEIMITTELS
ARIMIDEX 1 MG TABLETTEN MIT FILM .BESCHICHTET
02.0 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jede Filmtablette enthält 1 mg Anastrozol.
Hilfsstoffe mit bekannter Wirkung
Jede Filmtablette enthält 93 mg Lactose-Monohydrat (siehe Abschnitt 4.4).
Die vollständige Auflistung der sonstigen Bestandteile finden Sie in Abschnitt 6.1.
03.0 DARREICHUNGSFORM
Filmtablette.
Weiße, runde, bikonvexe Filmtabletten von ca. 6,1 mm mit der Prägung „A“ auf einer Seite und „Adx1“ auf der anderen Seite.
04.0 KLINISCHE INFORMATIONEN
04.1 Anwendungsgebiete
Arimidex ist indiziert in:
• Behandlung von hormonrezeptorpositivem fortgeschrittenem Brustkrebs bei postmenopausalen Frauen.
• Adjuvante Behandlung der frühen Stadien des hormonrezeptorpositiven invasiven Brustkrebses bei postmenopausalen Frauen.
• Adjuvante Behandlung von frühen Stadien von hormonrezeptorpositivem invasivem Brustkrebs bei postmenopausalen Frauen, die 2 oder 3 Jahre adjuvante Tamoxifen-Therapie erhalten haben.
04.2 Dosierung und Art der Anwendung
Dosierung
Die empfohlene Dosierung von Arimidex bei Erwachsenen einschließlich älterer Menschen beträgt einmal täglich eine 1-mg-Tablette.
Bei postmenopausalen Frauen mit hormonrezeptorpositivem frühinvasivem Brustkrebs beträgt die empfohlene Dauer der adjuvanten endokrinen Behandlung 5 Jahre.
Besondere Bevölkerungsgruppen
Kinder und Jugendliche
Arimidex wird bei Kindern und Jugendlichen aufgrund unzureichender Daten zur Sicherheit und Wirksamkeit nicht empfohlen (siehe Abschnitte 4.4 und 5.1).
Nierenversagen
Bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung werden keine Dosisanpassungen empfohlen. Bei Patienten mit schwerer Niereninsuffizienz sollte die Anwendung von Arimidex mit Vorsicht erfolgen (siehe Abschnitte 4.4 und 5.2).
Leberinsuffizienz
Bei Patienten mit leichter Leberfunktionsstörung werden keine Dosisanpassungen empfohlen. Bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung ist Vorsicht geboten (siehe Abschnitt 4.4).
Art der Verabreichung
Arimidex muss oral eingenommen werden.
04.3 Kontraindikationen
Arimidex ist kontraindiziert bei:
• Schwangerschaft oder bei stillenden Frauen.
• Patienten mit bekannter Überempfindlichkeit gegen Anastrozol oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
04.4 Besondere Warnhinweise und geeignete Vorsichtsmaßnahmen für die Anwendung
Im Allgemeinen
Arimidex sollte bei prämenopausalen Frauen nicht angewendet werden. Die Wechseljahre müssen biochemisch (mittels luteinisierendes Hormon [LH], follikelstimulierendes Hormon [FSH] und/oder Östradiolspiegel) bei jenen Patienten bestimmt werden, bei denen Zweifel über den Zustand der Wechseljahre bestehen Analoga.
Die gleichzeitige Anwendung von Arimidex mit Tamoxifen oder östrogenhaltigen Therapien sollte vermieden werden, da dies seine pharmakologische Wirkung verringern kann (siehe Abschnitte 4.5 und 5.1).
Wirkung auf die Knochenmineraldichte
Da Arimidex die zirkulierenden Östrogenspiegel senkt, kann es zu einer Abnahme der Knochenmineraldichte mit einem möglichen daraus resultierenden erhöhten Frakturrisiko führen (siehe Abschnitt 4.8).
Frauen mit Osteoporose oder Osteoporose-Risiko sollten zu Beginn der Behandlung und danach in regelmäßigen Abständen eine Untersuchung der Knochenmineraldichte durchführen.Eine Osteoporose-Behandlung oder -Prophylaxe sollte angemessen eingeleitet und engmaschig überwacht werden. Die Anwendung spezifischer Behandlungen, z. B. Bisphosphonate, kann in Betracht gezogen werden, da sie den weiteren durch Arimidex verursachten Verlust der Knochenmineraldichte bei postmenopausalen Frauen stoppen können (siehe Abschnitt 4.8).
Leberinsuffizienz
Arimidex wurde bei Brustkrebspatientinnen mit mittelschwerer oder schwerer Leberfunktionsstörung nicht untersucht. Die Anastrozol-Exposition kann bei Patienten mit Leberfunktionsstörung erhöht sein (siehe Abschnitt 5.2), die Anwendung von Arimidex bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung sollte mit Vorsicht erfolgen (siehe Abschnitt 4.2). jeden einzelnen Patienten.
Nierenversagen
Arimidex wurde bei Brustkrebspatientinnen mit schwerer Niereninsuffizienz nicht untersucht. Die Anastrozol-Exposition war bei Patienten mit schwerer Nierenfunktionsstörung (GRF .) nicht erhöht
Kinder und Jugendliche
Die Anwendung von Arimidex bei Mädchen und Jugendlichen wird nicht empfohlen, da die Sicherheit und Wirksamkeit bei dieser Patientengruppe nicht erwiesen ist (siehe Abschnitt 5.1).
Arimidex sollte bei männlichen Kindern mit Wachstumshormonmangel nicht zusätzlich zu einer Wachstumshormonbehandlung angewendet werden Die Wirksamkeit wurde in der zulassungsrelevanten klinischen Studie nicht nachgewiesen und die Sicherheit wurde nicht nachgewiesen (siehe Abschnitt 5.1). Da Anastrozol den Östradiolspiegel senkt, sollte Arimidex bei Mädchen mit Wachstumshormonmangel nicht zusätzlich zu einer Wachstumshormonbehandlung angewendet werden Langzeitdaten zur Sicherheit liegen bei pädiatrischen und jugendlichen Patienten nicht vor.
Überempfindlichkeit gegen Laktose
Dieses Produkt enthält Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, Lapp-Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
04.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Anastrozol hemmt CYPs, 1A2, 2C8 / 9 und 3A4 in vitro. Klinische Studien mit Antipyrin und Warfarin zeigen, dass Anastrozol 1 mg den Metabolismus von Antipyrin und R- und S-Warfarin nicht signifikant hemmt, was zeigt, dass die gleichzeitige Anwendung von Arimidex mit anderen Arzneimitteln klinisch unwahrscheinlich zu einer CYP-Enzym-vermittelten Wechselwirkung führt .
Die Enzyme, die den Metabolismus von Anastrozol vermitteln, wurden nicht identifiziert. Cimetidin, ein schwacher, unspezifischer Inhibitor von CYP-Enzymen, verändert die Plasmakonzentrationen von Anastrozol nicht. Die Wirkung potenter CYP-Inhibitoren ist nicht bekannt.
Eine Überprüfung der Sicherheitsdaten aus klinischen Studien ergab keine klinisch signifikanten Wechselwirkungen bei Patienten, die mit Arimidex und gleichzeitig mit anderen häufig verschriebenen Arzneimitteln behandelt wurden. Es gibt keine klinisch signifikanten Wechselwirkungen mit Bisphosphonaten (siehe Abschnitt 5.1).
Die gleichzeitige Anwendung von Arimidex mit Tamoxifen oder östrogenhaltigen Therapien sollte vermieden werden, da dies seine pharmakologische Wirkung verringern kann (siehe Abschnitte 4.4 und 5.1).
04.6 Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine Daten zur Anwendung von Arimidex bei schwangeren Frauen vor Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3) Arimidex ist während der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3).
Stillen
Es liegen keine Daten zur Anwendung von Arimidex während der Stillzeit vor. Arimidex ist bei stillenden Frauen kontraindiziert (siehe Abschnitt 4.3).
Fruchtbarkeit
Die Auswirkungen von Arimidex auf die menschliche Fertilität wurden nicht untersucht Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
04.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Arimidex hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Da jedoch bei der Anwendung von Arimidex über Asthenie und Schläfrigkeit berichtet wurde, ist beim Führen eines Fahrzeugs oder beim Bedienen von Maschinen Vorsicht geboten, wenn diese Symptome anhalten.
04.8 Nebenwirkungen
In der folgenden Tabelle sind Nebenwirkungen aus klinischen Studien, Studien nach Markteinführung oder Spontanberichten aufgeführt. Sofern nicht anders angegeben, wurden die folgenden Häufigkeitskategorien aus der Anzahl der berichteten Nebenwirkungen in einer großen Phase-III-Studie berechnet, die an 9.366 postmenopausalen Frauen mit operablem Brustkrebs unter adjuvanter Therapie über 5 Jahre durchgeführt wurde (Studie Arimidex, Tamoxifen, allein oder in Kombination [ATAC]).
Die nachfolgend aufgeführten Nebenwirkungen sind nach Häufigkeit und Organ- und Systemklasse (SOC) klassifiziert. Häufigkeitsklassen werden nach folgender Konvention definiert: sehr häufig (≥ 1/10), häufig (≥ 1/100 bis Kopfschmerzen, Hitzegefühl, Übelkeit, Hautausschlag, Arthralgie, Gelenkschmerzen/-steifheit, Arthritis und Asthenie).
* Karpaltunnelsyndrom-Ereignisse wurden bei Patienten, die Arimidex erhielten, in klinischen Studien bei mehr Patienten berichtet als bei Patienten, die Tamoxifen erhielten. Die meisten dieser Ereignisse traten jedoch bei Patienten mit identifizierbaren Risikofaktoren für die Entwicklung dieser Erkrankung auf.
** Da kutane Vaskulitis und Purpura Henoch-Schönlein in der ATAC-Studie nicht beobachtet wurden, kann die Häufigkeitskategorie für diese Ereignisse als „selten“ (≥ 0,01 % und
*** Vaginale Blutungen wurden insbesondere bei Patientinnen mit fortgeschrittenem Brustkrebs während der ersten Wochen nach Änderung einer bestehenden Hormontherapie während der Behandlung mit Arimidex häufig berichtet. Bei anhaltender Blutung sollte eine weitere Abklärung in Erwägung gezogen werden.
Die folgende Tabelle zeigt die Häufigkeit vordefinierter unerwünschter Ereignisse in der ATAC-Studie nach einer medianen Nachbeobachtungszeit von 68 Monaten, berichtet bei Patienten, die mit der Studientherapie behandelt wurden, und bis zu 14 Tage nach Absetzen der Studientherapie, unabhängig von der Kausalität.
Nach einer medianen Nachbeobachtungszeit von 68 Monaten wurden für die Arimidex- und Tamoxifen-Gruppen Frakturraten von 22 bzw. 15 pro 1.000 Patientenjahre beobachtet. Die beobachtete Frakturrate für Arimidex entspricht dem Bereich, der bei gleichaltrigen postmenopausalen Populationen berichtet wurde. Die Osteoporose-Inzidenz betrug 10,5 % bei den mit Arimidex behandelten Patienten und 7,3 % bei den mit Tamoxifen behandelten Patienten.
Es wurde nicht festgestellt, ob die in der ATAC-Studie bei mit Arimidex behandelten Patienten beobachteten Fraktur- und Osteoporoseraten eine protektive Wirkung von Tamoxifen oder eine spezifische Wirkung von Arimidex oder beides widerspiegeln.
Meldung von vermuteten Nebenwirkungen
Die Meldung vermuteter Nebenwirkungen, die nach der Zulassung des Arzneimittels aufgetreten sind, ist wichtig, da sie eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht. Angehörige von Gesundheitsberufen werden gebeten, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem zu melden. "Adresse https: //www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Überdosierung
Die klinische Erfahrung mit versehentlicher Überdosierung ist begrenzt.In Tierstudien hat Anastrozol eine geringe akute Toxizität gezeigt. Klinische Studien wurden mit verschiedenen Dosierungen von Arimidex durchgeführt, bis zu 60 mg als Einzeldosis bei gesunden männlichen Freiwilligen und bis zu 10 mg pro Tag bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs; diese Dosierungen wurden gut vertragen. Die Einzeldosis von Arimidex, die Symptome verursachen kann, die lebensbedrohlich sein können, wurde nicht ermittelt. Es gibt kein spezifisches Antidot gegen eine Überdosierung und die Behandlung sollte symptomatisch erfolgen.
Bei der Behandlung einer Überdosierung sollte die Möglichkeit in Betracht gezogen werden, dass mehrere Medikamente eingenommen wurden. Wenn die Person wachsam ist, kann Erbrechen induziert werden. Da Arimidex nicht stark an Plasmaproteine bindet, kann eine Dialyse hilfreich sein.Allgemeine unterstützende Maßnahmen sind angezeigt, einschließlich einer häufigen Überwachung der Vitalparameter und einer sorgfältigen Beobachtung des Patienten.
05.0 PHARMAKOLOGISCHE EIGENSCHAFTEN
05.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Enzymhemmer.
ATC-Code: L02B G03.
Wirkmechanismus und pharmakodynamische Wirkungen
Arimidex ist ein potenter und hochselektiver nichtsteroidaler Aromatasehemmer.Bei postmenopausalen Frauen wird Östradiol hauptsächlich im peripheren Gewebe produziert, nachdem Androstendion durch das Aromataseenzym in Östron umgewandelt wurde. Östron wird anschließend in Östradiol umgewandelt.Die Senkung der Plasmaspiegel von Östradiol hat sich bei Frauen mit Brustkrebs als günstigausgewirkt.Bei postmenopausalen Frauen führte Arimidex in einer Tagesdosis von 1 mg zu einer Suppression des Östradiolspiegels über 80 %, mit einem hochempfindlichen Test gemessen.
Arimidex besitzt keine progestogene, androgene oder östrogene Wirkung.
Tägliche Arimidex-Dosen von bis zu 10 mg zeigten keine Wirkung auf die Cortisol- oder Aldosteronsekretion, gemessen vor oder nach Standard-Stimulationstests mit adrenokortikotropem Hormon (ACTH), daher ist keine zusätzliche Gabe von Kortikosteroiden erforderlich.
Klinische Wirksamkeit und Sicherheit
Brustkrebs im fortgeschrittenen Stadium
Erstlinientherapie bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs
Zwei kontrollierte, doppelblinde klinische Studien mit ähnlichem experimentellem Design (Studie 1033IL / 0030 und Studie 1033IL / 0027) wurden durchgeführt, um die Wirksamkeit von Arimidex im Vergleich zu Tamoxifen als Erstlinientherapie bei postmenopausalen Frauen mit Brustkrebs zu bewerten lokal fortgeschritten mit positiven oder unbekannten Hormonrezeptoren Insgesamt 1.021 Patienten wurden randomisiert, um 1 mg Arimidex einmal täglich oder 20 mg Tamoxifen einmal täglich zu erhalten Die primären Endpunkte für beide Studien waren die Zeit bis zur Krankheitsprogression, die objektive Ansprechrate von Krankheit und Behandlungssicherheit.
Für die primären Endpunkte zeigte die Studie 1033IL / 0030 einen statistisch signifikanten Vorteil für Arimidex gegenüber Tamoxifen in Bezug auf die Krankheitsprogression (Hazard Ratio (HR) 1,42, 95 % Konfidenzintervall (KI) [1,11, 1,82], mediane Zeit bis zur Progression 11,1 und 5,6 Monate für Arimidex bzw. Tamoxifen, p = 0,006); die objektive Ansprechrate auf die Krankheit war sowohl für Arimidex als auch für Tamoxifen ähnlich. Studie 1033IL / 0027 zeigte, dass Arimidex und Tamoxifen eine ähnliche Ansprechrate und Zeit bis zum Fortschreiten der Krankheit aufwiesen. Die Ergebnisse der sekundären Endpunkte unterstützten die Ergebnisse der primären Wirksamkeitsziele. In den Behandlungsgruppen beider Studien traten nur wenige Todesfälle auf, sodass keine Rückschlüsse auf Unterschiede im Gesamtüberleben gezogen werden konnten.
Zweitlinientherapie bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs
Arimidex wurde in zwei kontrollierten klinischen Studien (Studie 0004 und Studie 0005) bei postmenopausalen Frauen mit fortgeschrittenem Brustkrebs untersucht, die nach einer Tamoxifen-Therapie sowohl bei fortgeschrittenem als auch bei frühem Krebs eine Progression aufwiesen. Insgesamt 764 Patienten wurden randomisiert und erhielten viermal täglich eine Einzeldosis von 1 mg oder 10 mg Arimidex oder 40 mg Megestrolacetat. Die primären Wirksamkeitsvariablen waren die Zeit bis zur Progression und die objektive Ansprechrate der Erkrankung. Außerdem wurden die Stabilitätsrate der Krankheit (mehr als 24 Wochen), die Progressionsrate und das Überleben berechnet. In beiden Studien gab es keine signifikanten Unterschiede zwischen den Behandlungsarmen in Bezug auf einen der Wirksamkeitsparameter.
Adjuvante Behandlung bei Patientinnen mit invasivem hormonrezeptorpositivem Brustkrebs im Frühstadium
In einer großen Phase-III-Studie an 9.366 postmenopausalen Frauen mit operablem Brustkrebs, die 5 Jahre lang behandelt wurden (siehe Tabelle unten), war Arimidex Tamoxifen im Hinblick auf das krankheitsfreie Überleben statistisch überlegen. Der beobachtete Vorteil des krankheitsfreien Überlebens war bei der prospektiv definierten hormonrezeptorpositiven Patientenpopulation für Arimidex größer als für Tamoxifen.
a Das krankheitsfreie Überleben umfasst alle Ereignisse vom Rezidivtyp und ist definiert als das erste Ereignis eines lokoregionalen Rezidivs, eines neuen kontralateralen Brustkrebses, eines Fernrezidivs oder eines Todes (aus jeder Ursache).
b Das krankheitsfreie Fernüberleben ist definiert als das erste Ereignis eines Fernrezidivs oder des Todes (aus welcher Ursache auch immer).
c Die Zeit bis zum Rezidiv ist definiert als das erste Ereignis eines lokoregionalen Rezidivs, eines neuen kontralateralen Brustkrebses, eines Fernrezidivs oder eines Brustkrebstodes.
d Die Zeit bis zum Fernrezidiv ist definiert als das erste Ereignis eines Fernrezidivs oder des Todes durch Brustkrebs.
e Anzahl (%) der verstorbenen Patienten.
Die Kombination von Arimidex und Tamoxifen zeigte bei allen Patienten, einschließlich derjenigen mit positivem Hormonrezeptor, keinen Wirksamkeitsvorteil im Vergleich zu Tamoxifen allein. Diese Behandlungsgruppe brach die Studie ab.
Bei einer aktualisierten medianen Nachbeobachtungszeit von 10 Jahren im Langzeitvergleich wurde gezeigt, dass die Wirkung der Behandlung mit Arimidex gegenüber Tamoxifen mit früheren Analysen übereinstimmt.
Adjuvante Behandlung von Brustkrebs im Frühstadium bei Hormonrezeptor-positiven Patientinnen, die eine adjuvante Tamoxifen-Behandlung erhalten
In einer Phase-III-Studie (Österreichische Studiengruppe Brust- und Darmkrebs [ABCSG] 8) bei 2.579 postmenopausalen Frauen mit frühem hormonrezeptorpositivem Brustkrebs, die sich einer Operation mit oder ohne Strahlentherapie und ohne Chemotherapie unterzogen hatten (siehe Tabelle unten), war der Ersatz von Arimidex nach 2 Jahren adjuvanter Behandlung mit Tamoxifen statistisch überlegen hinsichtlich des krankheitsfreien Überlebens nach einer medianen Nachbeobachtungszeit von 24 Monaten mit Tamoxifen fortzusetzen.
Diese Ergebnisse werden durch zwei weitere ähnliche Studien (GABG / ARNO 95 und ITA), in denen die Patienten operiert und Chemotherapie unterzogen wurden, sowie durch die kombinierte Auswertung der Studien ABCSG 8 und GABG / ARNO 95 gestützt.
Das Sicherheitsprofil von Arimidex in diesen 3 Studien entsprach dem Sicherheitsprofil, das zuvor bei postmenopausalen Frauen mit hormonrezeptorpositivem Brustkrebs im Frühstadium gefunden wurde.
Knochenmineraldichte (DMO)
In der Phase III / IV Studie (Untersuchung von Anastrozol mit dem Bisphosphonat Risedronat [SABER]), 234 postmenopausale Frauen mit hormonrezeptorpositiven Brustkrebskandidaten im Frühstadium für die Behandlung mit Arimidex 1 mg / Tag, wurden nach ihrem bestehenden Risiko für Fragilitätsfrakturen in Gruppen mit niedrigem, mittlerem und hohem Risiko eingeteilt. Der primäre Wirksamkeitsparameter war die Analyse der Knochenmassedichte der Wirbelsäule durch DEXA-Scans. Alle Patienten erhielten eine Behandlung mit Vitamin D und Kalzium. Patienten in der Niedrigrisikogruppe erhielten nur Arimidex (N = 42), die in der moderaten Gruppe wurden auf Arimidex® randomisiert plus Risedronat 35 mg einmal wöchentlich (n = 77) oder Arimidex plus Placebo (n = 77), und Hochrisikopatienten erhielten Arimidex plus Risedronat 35 mg einmal wöchentlich (n = 38) Der primäre Endpunkt war die Veränderung der Knochenmasse der Wirbelsäule vom Ausgangswert bis 12 Monate.
Die Hauptanalyse nach 12 Monaten zeigte, dass Patienten, die bereits ein mäßiges oder hohes Risiko für Fragilitätsfrakturen hatten, bei Behandlung mit Arimidex 1 mg / Tag keine Verringerung der Knochendichte (gemessen anhand der Knochenmineraldichte an der Wirbelsäule durch DEXA-Scans) aufwiesen in Kombination mit Risedronat 35 mg einmal wöchentlich Darüber hinaus wurde in der mit Arimidex 1 mg / Tag allein behandelten Niedrigrisikogruppe eine statistisch nicht signifikante Abnahme der Knochenmineraldichte (BMD) beobachtet Veränderung der Gesamt-BMD in der Hüfte gegenüber dem Ausgangswert nach 12 Monaten.
Diese Studie hebt hervor, dass die Verwendung von Bisphosphonaten bei der Behandlung eines möglichen Knochenmineralabbaus bei postmenopausalen Frauen mit Brustkrebs im Frühstadium in Erwartung einer Behandlung mit Arimidex in Betracht gezogen werden könnte.
Kinder und Jugendliche
Arimidex ist nicht zur Anwendung bei Kindern und Jugendlichen indiziert Die Wirksamkeit wurde bei der untersuchten pädiatrischen Population nicht nachgewiesen (siehe unten). Die Zahl der behandelten Probanden war zu gering, um verlässliche Schlussfolgerungen zur Sicherheit zu ziehen. Es liegen keine Daten zu potenziellen Langzeitwirkungen einer Behandlung mit Arimidex bei Kindern und Jugendlichen vor (siehe auch Abschnitt 5.3).
Die Europäische Arzneimittel-Agentur hat die Befreiung von der Pflicht zur Einreichung der Ergebnisse von Studien mit Arimidex in einer oder mehreren Untergruppen von pädiatrischen Populationen mit Kleinwuchs aufgrund von Wachstumshormonmangel (GHD), Testotoxikose, Gynäkomastie und McCune-Albright-Syndrom gewährt (siehe Abschnitt 4.2).
Kleinwuchs durch Wachstumshormonmangel
In einer randomisierten, doppelblinden, multizentrischen Studie wurden 52 pubertäre Männer (im Alter von 11-16 Jahren) mit GHD 12 bis 36 Monate lang mit Arimidex 1 mg / Tag oder Placebo in Kombination mit Wachstumshormon untersucht Arimidex hat 36 Monate abgeschlossen.
Bei wachstumsbezogenen Parametern wie geschätzter Erwachsenengröße, Körpergröße, SDS-Größe (Standardabweichungs-Score) und Wachstumsrate wurden im Vergleich zu Placebo keine statistisch signifikanten Unterschiede beobachtet. Endgültige Daten zur Körpergröße waren nicht verfügbar.Obwohl die Zahl der behandelten Kinder zu gering war, um verlässliche Schlussfolgerungen zur Sicherheit ziehen zu können, gab es in der Arimidex-Gruppe im Vergleich zu Placebo eine Zunahme der Frakturrate und einen Trend zu einer Abnahme der Knochenmineraldichte.
Testotoxikose
In einer offenen, nicht vergleichenden, multizentrischen Studie wurden 14 männliche Patienten (im Alter von 2 bis 9 Jahren) mit familiärer männlicher sexueller Frühreife, auch bekannt als Testotoxikose, in Kombination mit Arimidex und Bicalutamid behandelt. Das primäre Ziel war es, die Wirksamkeit und Sicherheit dieser Kombination während der 12 Monate zu überprüfen. Dreizehn der 14 eingeschlossenen Patienten beendeten eine 12-monatige kombinierte Behandlung (ein Patient verlor die Nachuntersuchung). Es gab keinen signifikanten Unterschied in der Wachstumsrate nach 12-monatiger Behandlung im Vergleich zur Wachstumsrate während der 6 Monate vor Studienbeginn.
Studien in Gynäkomastie
Studie 0006 war eine randomisierte, doppelblinde, multizentrische Studie, die an 82 Jungen im pubertären Alter (im Alter von 11 bis 18 Jahren) mit Gynäkomastie, die länger als 12 Monate andauerte, durchgeführt wurde und bis zu 6 Monate lang täglich mit Arimidex 1 mg/Tag oder Placebo behandelt wurde. Bei der Anzahl der Patientinnen, die nach 6-monatiger Behandlung eine Verringerung des Gesamtbrustvolumens um 50 % oder mehr aufwiesen, wurde kein signifikanter Unterschied zwischen der Arimidex 1 mg-Gruppe und der Placebo-Gruppe beobachtet.
Studie 0001 war eine offene pharmakokinetische Studie mit Mehrfachdosen von Arimidex 1 mg/Tag bei 36 pubertären Jungen mit Gynäkomastie von weniger als 12 Monaten Dauer. Sekundäre Ziele waren die Beurteilung des Anteils der Patientinnen, die eine Reduktion gegenüber dem Ausgangswert aufwiesen, das berechnete Gynäkomastievolumen beider Brüste kombiniert um mindestens 50 % zwischen dem ersten Tag und nach 6 Monaten der Behandlung sowie die Verträglichkeit und Patientensicherheit. Bei 56 % (20/36) der Jungen wurde nach 6 Monaten eine Verringerung des gesamten Brustvolumens um 50 % oder mehr beobachtet.
Studien zum McCune-Albright-Syndrom
Studie 0046 war eine internationale, multizentrische, explorative, offene Studie zu Arimidex bei 28 Mädchen (im Alter von 2 bis ≤ 10 Jahren) mit McCune-Albrigth-Syndrom (MAS). Das primäre Ziel war die Bewertung der Verträglichkeit und Wirksamkeit von Arimidex 1 mg / Tag bei Patienten mit MAS. Die Wirksamkeit der Studienbehandlung basierte auf dem Anteil der Patienten, die definierte Kriterien für vaginale Blutung, Knochenalter und Wachstumsrate erfüllten. Es wurde keine statistisch signifikante Veränderung der Häufigkeit von vaginalen Blutungen an Tagen während der Behandlung beobachtet. Es gab keine klinisch signifikanten Veränderungen im Tanner-Staging, mittleres Ovarialvolumen oder mittleres Uterusvolumen Es wurde kein statistisch signifikanter Unterschied in der Rate des Anstiegs des Knochenalters während der Behandlung gegenüber dem Ausgangswert beobachtet Wachstumsrate (in cm/Jahr) signifikant verringert (p
05.2 Pharmakokinetische Eigenschaften
Absorption
Anastrozol wird schnell resorbiert und maximale Plasmakonzentrationen werden im Allgemeinen innerhalb von zwei Stunden nach der Verabreichung (Nüchtern) erreicht. Nahrung verringert die Resorptionsrate leicht, aber nicht das Ausmaß der Resorption. Es wird angenommen, dass diese geringfügige Änderung der Resorptionsrate keine klinisch signifikante Wirkung hat auf die Steady-State-Plasmakonzentrationen bei einmal täglicher Gabe von Arimidex-Tabletten Nach 7 Tagen werden etwa 90-95 % der Steady-State-Plasmakonzentrationen von Anastrozol erreicht, und die Akkumulation erfolgte 3- bis 4-mal. Es gibt keine Hinweise auf eine Zeit- oder Dosisabhängigkeit der pharmakokinetischen Parameter von Anastrozol.
Die Pharmakokinetik von Anastrozol ist bei postmenopausalen Frauen altersunabhängig.
Verteilung
Anastrozol ist nur zu 40 % an Plasmaproteine gebunden.
Beseitigung
Anastrozol wird langsam mit einer Plasmaeliminationshalbwertszeit von 40 bis 50 Stunden eliminiert.
Anastrozol wird bei postmenopausalen Frauen weitgehend metabolisiert, wobei weniger als 10 % der Dosis innerhalb von 72 Stunden nach Einnahme unverändert über den Urin ausgeschieden werden. Der Metabolismus von Anastrozol erfolgt durch N-Dealkylierung, Hydroxylierung und Glucuronidierung. Die Metaboliten werden hauptsächlich über den Urin ausgeschieden Metabolit im Plasma, hemmt das Aromatase-Enzym nicht.
Nieren- oder Leberinsuffizienz
Die scheinbare Clearance (CL/F) von Anastrozol nach oraler Gabe war bei Probanden mit stabiler Leberzirrhose etwa 30 % niedriger als in der Kontrollgruppe (Studie 1033IL/0014). Die Plasmakonzentrationen von Anastrozol bei Freiwilligen mit Leberzirrhose blieben jedoch innerhalb des Konzentrationsbereichs, der bei gesunden Probanden in anderen Studien beobachtet wurde. Die Plasmakonzentrationen von Anastrozol, die in Langzeitstudien zur Wirksamkeit bei Patienten mit Leberinsuffizienz beobachtet wurden, blieben im Bereich der Plasmakonzentrationen von Anastrozol, die bei Patienten ohne Leberinsuffizienz beobachtet wurden.
Die scheinbare Clearance (CL/F) von Anastrozol nach oraler Gabe war bei Probanden mit schwerer Nierenfunktionsstörung (GFR .) nicht verändert
Kinder und Jugendliche
Bei Jungen mit pubertärer Gynäkomastie (10–17 Jahre) wurde Anastrozol mit einer Halbwertszeit von ca. 2 Tagen schnell resorbiert, breit verteilt und langsam eliminiert. Bei Mädchen (3–10 Jahre) war die Anastrozol-Clearance langsamer als bei Mädchen Jungen und die höchste Exposition. Bei Mädchen wurde Anastrozol weit verbreitet und langsam ausgeschieden.
05.3 Präklinische Sicherheitsdaten
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, kanzerogenem Potenzial, Reproduktions- und Entwicklungstoxizität für die vorhergesagte Population lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Akute Toxizität
In Tierstudien wurde Toxizität nur bei hohen Dosen beobachtet. In Studien zur akuten Toxizität an Nagetieren war die mediane letale Dosis von Anastrozol oral über 100 mg/kg/Tag und intraperitoneal über 50 mg/kg/Tag. In einer Studie zur akuten Toxizität bei Hunden betrug die mediane letale Dosis mehr als 45 mg/kg/Tag oral.
Chronische Toxizität
In Tierstudien wurden Nebenwirkungen nur bei hohen Dosen beobachtet. Toxizitätsstudien mit Mehrfachdosierung wurden sowohl an Ratten als auch an Hunden durchgeführt. In diesen Studien wurden keine wirkungslosen Dosen festgestellt; Die bei niedrigen Dosen (1 mg / kg / Tag) und mittleren Dosen (Hund 3 mg / kg / Tag; Ratte 5 mg / kg / Tag) beobachteten Wirkungen standen jedoch sowohl mit den pharmakologischen als auch enzyminduzierenden Eigenschaften von Anastrozol und sie waren nicht mit signifikanten toxikologischen oder degenerativen Veränderungen verbunden.
Mutagenese
Studien zur genetischen Toxizität mit Anastrozol zeigten, dass das Produkt weder mutagen noch klastogen ist.
Reproduktionstoxizität
In einer Fertilitätsstudie wurden frisch abgesetzten männlichen Ratten 10 Wochen lang orale Dosen von 50 oder 400 mg/l Anastrozol über das Trinkwasser verabreicht.Die mittleren Plasmakonzentrationen betrugen 44,4 (± 14,7) ng/ml bzw. 165 (± 90) ng/ml. Die Reproduktionsindizes wurden in beiden Dosisgruppen beeinträchtigt, während eine Verringerung der Fertilität nur bei der Dosis von 400 mg/l erkennbar war. Die Reduktion war vorübergehend, da alle Reproduktions- und Fertilitätsparameter nach einer medikamentenfreien Erholungsphase von 9 Wochen den Werten der Kontrollgruppe ähnlich waren.
Die orale Verabreichung von Anastrozol 1 mg / kg / Tag an weibliche Ratten führte zu einer hohen Infertilität und bei einer Dosis von 0,02 mg / kg / Tag zu einer Zunahme des Präimplantationsverlusts. Diese Effekte traten bei klinisch relevanten Dosen auf Menschen sind nicht auszuschließen. Diese Wirkungen standen im Zusammenhang mit den pharmakologischen Wirkungen des Tierarzneimittels und gingen nach einer 5-wöchigen Wartezeit vollständig zurück.
Die orale Verabreichung von Anastrozol an trächtige Ratten und Kaninchen verursachte bei Dosen bis zu 1 bzw. 0,2 mg / kg / Tag keine teratogenen Wirkungen. Die beobachteten Wirkungen (wie Plazentavergrößerung bei Ratten und Schwangerschaftsabbruch bei Kaninchen) standen im Zusammenhang mit den pharmakologischen Wirkungen des Tierarzneimittels.
Das Überleben der Nachkommen weiblicher Ratten, die mit Anastrozol in Dosen von 0,02 mg/kg/Tag oder mehr (vom 17. Tag der Trächtigkeit bis zum 22. Tag nach der Geburt) behandelt wurden, war beeinträchtigt. Diese Wirkungen standen im Zusammenhang mit den pharmakologischen Wirkungen des Produkts bei der Lieferung. Es gab keine nachteiligen Auswirkungen auf das Verhalten oder die Fortpflanzungsleistung der Nachkommen der ersten Generation, die auf eine mütterliche Behandlung mit Anastrozol zurückzuführen waren.
Karzinogenese
Eine zweijährige Kanzerogenitätsstudie an Ratten zeigte nur bei der hohen Dosis (25 mg / kg / Tag) eine erhöhte Inzidenz von Leberneoplasmen und Uterusstromapolypen bei Frauen und Schilddrüsenadenomen bei Männern. Diese Veränderungen traten bei einer Dosis auf, die einer 100-fach höheren Exposition ausgesetzt als die, die bei therapeutischen Dosen beim Menschen auftritt, und werden für die Behandlung von Patienten mit Anastrozol nicht als klinisch relevant erachtet.
Eine zweijährige Karzinogenitätsstudie an Mäusen zeigte die Induktion von gutartigen Ovarialtumoren und eine "veränderte" Inzidenz von lymphoretikulären Neoplasien (weniger histiozytäre Sarkome bei Frauen und mehr Todesfälle durch Lymphome). Diese Veränderungen gelten als speziesspezifisch für die Aromatasehemmung bei Mäusen und werden für die Behandlung von Patienten mit Anastrozol nicht als klinisch relevant erachtet.
06.0 PHARMAZEUTISCHE INFORMATIONEN
06.1 Hilfsstoffe
Lactose-Monohydrat
Povidon
Natriumstärkeglykolat
Magnesiumstearat
Hypromellose
Macrogol 300
Titandioxid
06.2 Inkompatibilität
Nicht relevant.
06.3 Gültigkeitsdauer
5 Jahre.
06.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Nicht über 30 °C lagern.
06.5 Art der unmittelbaren Verpackung und Inhalt des Packstücks
PVC/Aluminium-Blister. Packungen mit 20, 28, 30, 84, 98, 100, 300 Tabletten in einer Schachtel. Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
06.6 Gebrauchs- und Handhabungshinweise
Nicht verwendete Arzneimittel und Abfälle aus diesem Arzneimittel müssen gemäß den örtlichen Vorschriften entsorgt werden.
07.0 INHABER DER MARKETING-ERLAUBNIS
AstraZeneca UK Limited
600 Fähigkeit Grün
Luton LU1 3LU
Vereinigtes Königreich
Vertreter für Italien:
AstraZeneca S.p.A.,
Volta-Palast,
Via F. Sforza,
Basiglio (MI)
08.0 NUMMER DER MARKETING-ERLAUBNIS
031809041 1 mg Filmtabletten 20 Tabletten
031809015 1 mg Filmtabletten 28 Tabletten
031809027 1 mg Filmtabletten 30 Tabletten
031809039 1 mg Filmtabletten 84 Tabletten
031809054 1 mg Filmtabletten 100 Tabletten
031809066 1 mg Filmtabletten 300 Tabletten
09.0 DATUM DER ERSTEN GENEHMIGUNG ODER ERNEUERUNG DER GENEHMIGUNG
20 Tabletten von 1 mg:
Datum A.I.C. 11.12.97 / Erneuerungsdatum: 12. Mai 2015
28 Tabletten von 1 mg:
Datum A.I.C. 23.05.96 / Erneuerungsdatum: 12. Mai 2015
30 Tabletten von 1 mg:
Datum A.I.C. 11.12.97 / Erneuerungsdatum: 12. Mai 2015
84 Tabletten von 1 mg:
Datum A.I.C. 11.12.97 / Erneuerungsdatum: 12. Mai 2015
100 Tabletten von 1 mg:
Datum A.I.C. 11.12.97 / Erneuerungsdatum: 12. Mai 2015
300 Tabletten von 1 mg:
Datum A.I.C. 11.12.97 / Erneuerungsdatum: 12. Mai 2015
10.0 DATUM DER ÜBERARBEITUNG DES TEXTs
12. Mai 2015