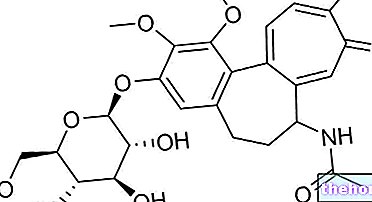
Wirkstoffe: Ulipristal (Ulipristalacetat)
Esmya 5 mg Tabletten
Warum wird Esmya verwendet? Wofür ist das?
Esmya enthält den Wirkstoff Ulipristalacetat. Es wird verwendet, um mittelschwere bis schwere Symptome von Uterusmyomen (auch Myome genannt) zu behandeln, bei denen es sich um gutartige Tumoren der Gebärmutter handelt.
Esmya wird bei erwachsenen Frauen (über 18 Jahren) angewendet, die die Menopause noch nicht erreicht haben.
Bei manchen Frauen können Uterusmyome starke Menstruationsblutungen ("Menstruation"), Beckenschmerzen (Beschwerden im Unterbauch) und Druck auf andere Organe verursachen.
Dieses Arzneimittel wirkt, indem es die Aktivität von Progesteron, einem natürlich vorkommenden Hormon im Körper, modifiziert. Es wird vor einer Operation zur Entfernung von Myomen oder zur Langzeitbehandlung von Myomen angewendet, um ihre Größe zu reduzieren, Blutungen zu stoppen oder zu reduzieren und die Anzahl der roten Blutkörperchen zu erhöhen.
Kontraindikationen Wann Esmya nicht angewendet werden sollte
Sie sollten sich bewusst sein, dass die meisten Frauen während der Behandlung und einige Wochen danach keine Menstruationsblutung (Menstruation) haben.
Nehmen Sie Esmya nicht ein
- wenn Sie allergisch gegen Ulipristalacetat oder einen der sonstigen Bestandteile von Esmya . sind
- wenn Sie schwanger sind oder stillen;
- wenn Sie vaginale Blutungen haben, die nicht durch Uterusmyome verursacht wurden;
- wenn Sie Krebs der Gebärmutter, des Gebärmutterhalses (des Gebärmutterhalses), der Eierstöcke oder der Brüste haben.
Vorsichtsmaßnahmen für die Anwendung Was sollten Sie vor der Einnahme von Esmya beachten?
- Wenn Sie ein hormonelles Kontrazeptivum (wie die Pille) einnehmen (siehe „Einnahme von Esmya mit anderen Arzneimitteln“), müssen Sie während der Einnahme von Esmya eine zuverlässige alternative Verhütungsmethode (wie ein Kondom) anwenden.
- Wenn Sie eine Leber- oder Nierenerkrankung haben, informieren Sie Ihren Arzt oder Apotheker, bevor Sie Esmya einnehmen.
- Wenn Sie schweres Asthma haben, ist Esmya möglicherweise nicht für Sie geeignet. Besprechen Sie dies mit Ihrem Arzt.
Die Behandlung mit Esmya führt im Allgemeinen zu einer signifikanten Verringerung der Menstruationsblutung (Menstruation) oder kann diese sogar innerhalb der ersten 10 Tage der Behandlung stoppen. Wenn Sie jedoch weiterhin übermäßige Blutungen haben, informieren Sie Ihren Arzt.
Die Menstruation wird normalerweise innerhalb von 4 Wochen nach Beendigung der Behandlung mit Esmya wieder aufgenommen. Die Gebärmutterschleimhaut kann sich infolge der Behandlung mit Esmya verdicken oder verändern.Diese Veränderungen verschwinden nach Beendigung der Behandlung und Wiederaufnahme der Menstruation.
Kinder und Jugendliche
Kinder unter 18 Jahren sollten Esmya nicht einnehmen.
Wechselwirkungen Welche Medikamente oder Lebensmittel können die Wirkung von Esmya® verändern?
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen, kürzlich andere Arzneimittel eingenommen haben oder beabsichtigen andere Arzneimittel einzunehmen.
Wenn Sie eines der unten aufgeführten Arzneimittel einnehmen, informieren Sie Ihren Arzt oder Apotheker, da diese Arzneimittel mit Esmya interagieren können:
- Einige Arzneimittel zur Behandlung von Herzproblemen (z. B. Digoxin).
- Einige Arzneimittel zur Vorbeugung von Schlaganfällen und Blutgerinnseln (z. B. Dabigatranethexylat).
- Einige Arzneimittel zur Behandlung von Epilepsie (zB Phenytoin, Fosphenytoin, Phenobarbital, Carbamazepin, Oxcarbazepin, Primidon).
- Einige Arzneimittel zur Behandlung einer HIV-Infektion (zB Ritonavir, Efavirenz, Nevirapin).
- Arzneimittel zur Behandlung einiger bakterieller Infektionen (z. B. Rifampicin, Telithromycin, Clarithromycin, Erythromycin, Rifabutin).
- Einige Arzneimittel zur Behandlung von Pilzinfektionen (z. B. Ketoconazol (außer Shampoo), Itraconazol).
- Pflanzliche Heilmittel, die Johanniskraut (Hypericum perforatum) enthalten, zur Behandlung von Depressionen oder Angstzuständen.
- Einige Arzneimittel zur Behandlung von Depressionen (z. B. Nefazodon).
- Einige Arzneimittel zur Behandlung von Bluthochdruck (zB Verapamil).
Esmya verringert wahrscheinlich die Wirksamkeit einiger hormoneller Kontrazeptiva Auch hormonelle Kontrazeptiva und Gestagene (zB Norethindron oder Levonorgestrel) können die Wirksamkeit von Esmya verringern. Folglich werden hormonelle Kontrazeptiva nicht empfohlen und Sie müssen während der Behandlung mit Esmya eine zuverlässige alternative Verhütungsmethode wie ein Kondom anwenden.
Einnahme von Esmya zusammen mit Nahrungsmitteln und Getränken
Sie sollten während der Einnahme von Esmya keinen Grapefruitsaft trinken.
Warnungen Es ist wichtig zu wissen, dass:
Schwangerschaft und Stillzeit
Nehmen Sie Esmya nicht ein, wenn Sie schwanger sind. Die Behandlung während der Schwangerschaft kann den Verlauf beeinflussen (wir wissen nicht, ob Esmya dem Fötus schaden oder eine Fehlgeburt verursachen kann). Wenn Sie während der Einnahme von Esmya schwanger werden, müssen Sie die Einnahme von Esmya unverzüglich beenden und Ihren Arzt oder Apotheker kontaktieren.
Esmya verringert wahrscheinlich die Wirksamkeit einiger hormoneller Kontrazeptiva (siehe „Einnahme von Esmya mit anderen Arzneimitteln"). Esmya geht in die Muttermilch über. Sie sollten daher während der Einnahme von Esmya nicht stillen.
Fragen Sie vor der Einnahme von Arzneimitteln Ihren Arzt oder Apotheker um Rat.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Esmya kann leichten Schwindel verursachen (siehe Abschnitt 4 „Mögliche Nebenwirkungen“). Wenn Sie diese Symptome bemerken, dürfen Sie kein Fahrzeug führen und keine Maschinen bedienen.
Dosis, Methode und Zeitpunkt der Verabreichung Wie ist Esmya anzuwenden: Dosierung
Nehmen Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt ein. Fragen Sie im Zweifelsfall Ihren Arzt oder Apotheker.
Die empfohlene Dosis beträgt eine 5-mg-Tablette pro Tag für Behandlungszyklen von jeweils bis zu 3 Monaten. Wenn Ihnen mehrere 3-monatige Behandlungen mit Esmya verordnet wurden, sollten Sie mit jeder Behandlung so bald wie möglich während des zweiten Menstruationszyklus nach Abschluss der vorherigen Behandlung beginnen.
Sie sollten mit der Einnahme von Esmya immer in der ersten Woche Ihres Menstruationszyklus beginnen.
Die Tablette sollte mit Wasser geschluckt werden und kann mit oder ohne Nahrung eingenommen werden.
Überdosierung Was ist zu tun, wenn Sie zu viel Esmya eingenommen haben?
Wenn Sie eine größere Menge von Esmya eingenommen haben, als Sie sollten
Die Erfahrungen mit der gleichzeitigen Einnahme mehrerer Dosen Esmya sind begrenzt. Bei gleichzeitiger Einnahme mehrerer Dosen dieses Arzneimittels wurden keine schwerwiegenden schädlichen Wirkungen berichtet. Wenn Sie jedoch eine größere Menge von Esmya eingenommen haben, als Sie sollten, wird empfohlen, dass Sie Ihren Arzt oder Apotheker konsultieren.
Wenn Sie die Einnahme von Esmya vergessen haben
Wenn Sie eine Dosis vergessen haben, die Sie vor weniger als 12 Stunden einnehmen sollten, nehmen Sie diese ein, sobald Sie dies bemerken. Wenn mehr als 12 Stunden vergangen sind, lassen Sie die vergessene Dosis aus und nehmen Sie wie gewohnt nur eine Tablette ein Nehmen Sie nicht die doppelte Dosis ein, wenn Sie die vergessene Tablette vergessen haben.
Wenn Sie die Einnahme von Esmya abbrechen
Esmya sollte während Behandlungszyklen von bis zu 3 Monaten täglich eingenommen werden. Brechen Sie die Einnahme der Tabletten während jeder Behandlungsphase nicht ohne den Rat Ihres Arztes ab, auch wenn Sie sich besser fühlen, da die Symptome später wieder auftreten können.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Nebenwirkungen Was sind die Nebenwirkungen von Esmya
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Sehr häufige Nebenwirkungen (betreffen mehr als 1 von 10 Behandelten):
- Verringerung oder Ausbleiben von Menstruationsblutungen (Amenorrhoe)
- Verdickung der Gebärmutterschleimhaut (Verdickung der Gebärmutterschleimhaut)
Häufige Nebenwirkungen (betreffen bis zu 1 von 10 Behandelten):
- Kopfschmerzen
- Schwindelgefühl (Schwindel)
- Bauchschmerzen, Übelkeit (Übelkeit)
- Akne
- Schmerzen in Muskeln und Knochen (muskuloskelettale)
- Flüssigkeitssack in den Eierstöcken (Ovarialzyste), Brustspannung / Schmerzen, Schmerzen im Unterbauch (Becken)
- Hitzewallungen
- Müdigkeit (Müdigkeit)
- Gewichtszunahme.
Gelegentliche Nebenwirkungen (betreffen bis zu 1 von 100 Behandelten):
- Angst
- Stimmungsschwankungen
- Schwindel
- Mundtrockenheit, Verstopfung
- Haarausfall, trockene Haut, vermehrtes Schwitzen
- Rückenschmerzen
- Urinverlust
- Blutungen der Gebärmutter (Uterusblutung), Ausfluss aus der Scheide, abnormale Blutungen aus der Scheide, Brustbeschwerden
- Schwellung durch Wassereinlagerungen (Ödeme)
- extreme Müdigkeit (Asthenie)
- durch Tests nachgewiesener Anstieg des Blutcholesterins, durch Tests nachgewiesener Anstieg der Blutfette (Triglyceride).
Seltene Nebenwirkungen (betreffen bis zu 1 von 1.000 Behandelten):
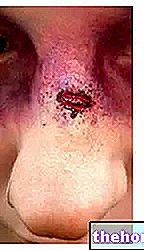
- Nasenbluten
- Verdauungsstörungen, Blähungen
- Ruptur des Flüssigkeitssacks in den Eierstöcken (Ruptur der Eierstockzyste)
- Brustschwellung.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker, einschließlich aller möglichen Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem melden. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
Ablauf und Aufbewahrung
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Sie dürfen dieses Arzneimittel nicht nach dem Verfallsdatum verwenden, das auf der Packung und der Blisterpackung nach „EXP“ angegeben ist. Das Ablaufdatum bezieht sich auf den letzten Tag dieses Monats.
Bewahren Sie die Blisterpackung im Umkarton auf, um das Arzneimittel vor Licht zu schützen.
Werfen Sie Arzneimittel nicht in das Abwasser oder den Hausmüll. Fragen Sie Ihren Apotheker, wie Sie Arzneimittel, die Sie nicht mehr verwenden, entsorgen. Dies trägt zum Schutz der Umwelt bei.
Was Esmya enthält
- Der Wirkstoff ist Ulipristalacetat. Eine Tablette enthält 5 mg Ulipristalacetat.
- Die sonstigen Bestandteile sind mikrokristalline Cellulose, Mannit, Croscarmellose-Natrium, Talkum und Magnesiumstearat.
Beschreibung wie Esmya aussieht und Inhalt der Packung
Esmya ist eine weiße bis cremefarbene, gebogene 7 mm runde Tablette mit der Prägung „ES5“ auf einer Seite.
Esmya ist in Al / PVC / PE / PVDC-Blisterpackungen in Kartons mit 28, 30 und 84 Tabletten oder in Al / PVC / PVDC-Blisterpackungen in Kartons mit 28 und 84 Tabletten erhältlich.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Quelle Packungsbeilage: AIFA (Italienische Arzneimittelbehörde). Im Januar 2016 veröffentlichter Inhalt. Die vorliegenden Informationen können nicht aktuell sein.
Um Zugriff auf die aktuellste Version zu erhalten, ist es ratsam, auf die Website der AIFA (Italienische Arzneimittelbehörde) zuzugreifen. Haftungsausschluss und nützliche Informationen.
01.0 BEZEICHNUNG DES ARZNEIMITTELS
ESMYA 5 MG TABLETTEN
02.0 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Tablette enthält 5 mg Ulipristalacetat.
Die vollständige Auflistung der sonstigen Bestandteile finden Sie in Abschnitt 6.1.
03.0 DARREICHUNGSFORM
Tablette.
Runde, weiße bis cremefarbene, bikonvexe 7-mm-Tablette mit der Prägung „ES5“ auf einer Seite.
04.0 KLINISCHE INFORMATIONEN
04.1 Anwendungsgebiete
Ulipristalacetat ist angezeigt zur präoperativen Behandlung mittelschwerer bis schwerer Symptome von Uterusmyomen bei erwachsenen Frauen im gebärfähigen Alter.
Ulipristalacetat ist angezeigt zur intermittierenden Behandlung mittelschwerer bis schwerer Symptome von Uterusmyomen bei erwachsenen Frauen im gebärfähigen Alter.
04.2 Dosierung und Art der Anwendung
Dosierung
Die Behandlung besteht aus einer einmal täglich einzunehmenden 5-mg-Tablette für Behandlungszyklen von jeweils bis zu 3 Monaten.
Behandlungen sollten erst beginnen, wenn die Menstruation eingetreten ist:
- Der erste Behandlungszyklus sollte in der ersten Woche der Menstruation beginnen.
- Folgekurse sollten so früh wie möglich in der ersten Woche der zweiten Menstruation nach Abschluss des vorherigen Behandlungszyklus beginnen.
Der behandelnde Arzt muss dem Patienten erklären, dass die Abbruchintervalle der Behandlung eingehalten werden müssen.
Die intermittierende wiederholte Behandlung wurde für bis zu 4 intermittierende Behandlungszyklen untersucht.
Wenn eine Patientin die Einnahme einer Dosis vergisst, sollte sie so schnell wie möglich Ulipristalacetat einnehmen. Wenn seit der vergessenen Dosis mehr als 12 Stunden vergangen sind, sollte die Patientin die vergessene Dosis nicht mehr einnehmen, sondern einfach ihr gewohntes Dosierungsschema wieder aufnehmen.
Besondere Bevölkerungsgruppen
Nierenversagen
Bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung wird keine Dosisanpassung empfohlen. Da keine spezifischen Studien vorliegen, wird Ulipristalacetat bei Patienten mit schwerer Nierenfunktionsstörung nicht empfohlen, es sei denn, der Patient wird engmaschig überwacht (siehe Abschnitte 4.4 und 5.2).
Leberinsuffizienz
Bei Patienten mit leichter Leberfunktionsstörung wird keine Dosisanpassung empfohlen. Da keine spezifischen Studien vorliegen, wird Ulipristalacetat bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung nicht empfohlen, es sei denn, der Patient wird engmaschig überwacht (siehe Abschnitte 4.4 und 5.2).
Kinder und Jugendliche
Es gibt keine Indikation für eine spezifische Anwendung von Ulipristalacetat bei Kindern und Jugendlichen. Die Sicherheit und Wirksamkeit von Ulipristalacetat wurde nur bei Frauen im Alter von mindestens 18 Jahren bestimmt.
Art der Verabreichung
Die Tabletten können mit oder ohne Nahrung eingenommen werden.
04.3 Kontraindikationen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Schwangerschaft und Stillzeit.
Vaginale Blutungen unbekannter Ätiologie oder aus anderen Gründen als dem Uterusmyom.
Gebärmutter-, Gebärmutterhals-, Eierstock- oder Brustkrebs.
04.4 Besondere Warnhinweise und geeignete Vorsichtsmaßnahmen für die Anwendung
Ulipristalacetat sollte nur nach sorgfältiger Diagnose verschrieben werden. Vor der Behandlung muss eine Schwangerschaft ausgeschlossen werden. Führen Sie bei Verdacht auf eine Schwangerschaft einen Schwangerschaftstest durch, bevor Sie eine neue Behandlung beginnen.
Empfängnisverhütung
Die gleichzeitige Anwendung eines Gestagen-freisetzenden Intrauterinpessars, eines Gestagen-freisetzenden Intrauterinpessars oder einer kombinierten oralen Antibabypille wird nicht empfohlen (siehe Abschnitt 4.5).Obwohl die meisten Frauen, die eine therapeutische Dosis von Ulipristalacetat einnehmen, eine Anovulation erleiden, wird während der Behandlung empfohlen: die Anwendung einer nicht-hormonellen Verhütungsmethode.
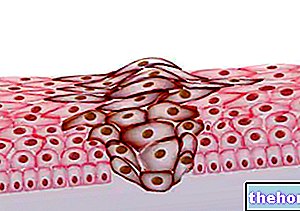
Endometriumveränderungen
Ulipristalacetat übt eine spezifische pharmakodynamische Wirkung auf das Endometrium aus:
Bei mit Ulipristalacetat behandelten Patienten können Veränderungen der Histologie des Endometriums beobachtet werden. Diese Veränderungen sind nach Beendigung der Behandlung reversibel.
Diese histologischen Veränderungen werden als „Progesteron-Rezeptor-Modulator-assoziierte Endometriumveränderungen“ (PAEC) bezeichnet und sollten nicht mit Endometriumhyperplasie verwechselt werden (siehe Abschnitte 4.8 und 5.1).
Im Verlauf der Behandlung kann es auch zu einer reversiblen Dickenzunahme des Endometriums kommen.
Bei wiederholter intermittierender Behandlung wird eine regelmäßige Überwachung des Endometriums empfohlen, einschließlich einer jährlichen Ultraschalluntersuchung, die nach Wiederaufnahme der Menstruation während der Behandlungsunterbrechung durchgeführt wird.
Wenn eine Verdickung des Endometriums besteht, die nach Wiederaufnahme der Menstruation während der Behandlungsabsetzzeiten oder für mehr als 3 Monate nach Beendigung der Behandlungszyklen anhält und / oder ein verändertes Blutungsprofil beobachtet wird (siehe "Blutungsprofil") , sollten Untersuchungen einschließlich einer Endometriumbiopsie durchgeführt werden, um andere zugrunde liegende Erkrankungen, einschließlich maligner Endometriumkarzinome, auszuschließen.
Bei Hyperplasie (ohne Atypie) wird eine Überwachung gemäß der üblichen klinischen Praxis empfohlen (z. B. Nachkontrolle nach 3 Monaten). Bei atypischer Hyperplasie müssen die in der üblichen klinischen Praxis erforderlichen Untersuchungen und Verfahren durchgeführt werden.
Behandlungszyklen sollten jeweils 3 Monate nicht überschreiten, da das Risiko von Nebenwirkungen auf das Endometrium bei Fortführung ohne Behandlungsunterbrechung nicht bekannt ist.
Blutungsprofil
Die Patienten sollten darauf hingewiesen werden, dass die Behandlung mit Ulipristalacetat im Allgemeinen innerhalb der ersten 10 Behandlungstage zu einer signifikanten Verringerung des menstruellen Blutverlusts oder der Amenorrhoe führt. Bei anhaltenden starken Blutungen sollte die Patientin ihren Arzt informieren. Die Menstruation wird im Allgemeinen innerhalb von 4 Wochen nach dem Ende jedes Behandlungszyklus wieder auftreten.
Wenn bei wiederholter intermittierender Behandlung nach anfänglicher Blutungsreduktion oder Amenorrhoe ein anhaltendes oder unerwartet verändertes Blutungsmuster, wie z. einschließlich Malignität des Endometriums.
Die intermittierende wiederholte Behandlung wurde für bis zu 4 intermittierende Behandlungszyklen untersucht.
Nierenversagen
Es ist nicht zu erwarten, dass eine Niereninsuffizienz die Elimination von Ulipristalacetat signifikant verändert. Da keine spezifischen Studien vorliegen, wird Ulipristalacetat bei Patienten mit schwerer Nierenfunktionsstörung nicht empfohlen, es sei denn, der Patient wird streng überwacht (siehe Abschnitt 4.2).
Leberinsuffizienz
Es liegen keine therapeutischen Erfahrungen mit Ulipristalacetat bei Patienten mit Leberinsuffizienz vor. Es ist zu erwarten, dass eine Leberinsuffizienz die Elimination von Ulipristalacetat verändert, was zu einer erhöhten Exposition führt (siehe Abschnitt 5.2).Dieser Effekt wird bei Patienten mit leichter Leberfunktionsstörung als nicht klinisch relevant angesehen Die Anwendung von Ulipristalacetat bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung, es sei denn der Patient wird engmaschig überwacht (siehe Abschnitt 4.2).
Gleichzeitige Behandlungen
Die gleichzeitige Anwendung von moderaten (z. B. Erythromycin, Grapefruitsaft, Verapamil) oder starken (z. B. Ketoconazol, Ritonavir, Nefazodon, Itraconazol, Telithromycin, Clarithromycin) Inhibitoren von CYP3A4 und Ulipristalacetat wird nicht empfohlen (siehe Abschnitt 4.5).
Gleichzeitige Anwendung von Ulipristalacetat und starken CYP3A4-Induktoren (z. B. Rifampicin, Rifabutin, Carbamazepin, Oxcarbazepin, Phenytoin, Fosphenytoin, Phenobarbital, Primidon, Johanniskraut, Efavirenz, Nevirapin, Ritonavir wird nicht empfohlen, siehe Abschnitt 4.5). ).
Asthmatiker
Die Anwendung bei Frauen mit schwerem Asthma, das durch orale Glukokortikoide nicht ausreichend kontrolliert wird, wird nicht empfohlen.
04.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Mögliche Beeinträchtigung von Ulipristalacetat durch andere Arzneimittel:
Hormonelle Verhütungsmittel
Ulipristalacetat hat eine Steroidstruktur und wirkt als selektiver Progesteronrezeptor-Modulator mit überwiegend inhibitorischer Wirkung auf den Progesteronrezeptor. Daher ist es wahrscheinlich, dass hormonelle und Gestagen-Kontrazeptiva die Wirksamkeit von Ulipristalacetat aufgrund einer kompetitiven Wirkung auf den Progesteronrezeptor verringern. Die gleichzeitige Anwendung von gestagenhaltigen Arzneimitteln wird daher nicht empfohlen (siehe Abschnitte 4.4 und 4.6).
CYP3A4-Inhibitoren
Nach Verabreichung des mäßigen CYP3A4-Inhibitors Erythromycinpropionat (500 mg zweimal täglich über 9 Tage) an gesunde Probanden stiegen die Cmax und die AUC von Ulipristalacetat um das 1,2- bzw. 2,9-Fache, des aktiven Metaboliten von Ulipristalacetat um das 1,5-Fache, während die Cmax des aktiven Metaboliten nahm ab (0,52-fache Änderung).
Nach Verabreichung des starken CYP3A4-Inhibitors Ketoconazol (400 mg einmal täglich über 7 Tage) an gesunde Freiwillige stiegen Cmax und AUC von Ulipristalacetat um das 2-Fache bzw. während die Cmax des aktiven Metaboliten abnahm (0,53-fache Veränderung).
Bei der Anwendung von Ulipristalacetat an Patienten, die gleichzeitig leichte CYP3A4-Inhibitoren erhalten, ist keine Dosisanpassung erforderlich. Die gleichzeitige Anwendung von mäßigen oder starken CYP3A4-Inhibitoren und Ulipristalacetat wird nicht empfohlen (siehe Abschnitt 4.4).
CYP3A4-Induktoren
Die Verabreichung des starken CYP3A4-Induktors Rifampicin (300 mg zweimal täglich für 9 Tage) an gesunde Probanden reduzierte Cmax und AUC von Ulipristalacetat und seinem aktiven Metaboliten signifikant um 90 % oder mehr und verkürzte die Halbwertszeit von Ulipristalacetat um das 2,2-Fache Die gleichzeitige Anwendung von Ulipristalacetat und starken CYP3A4-Induktoren (z.Rifampicin, Rifabutin, Carbamazepin, Oxcarbazepin, Phenytoin, Fosphenytoin, Phenobarbital, Primidon, Johanniskraut, Efavirenz, Nevirapin, Langzeitanwendung von Ritonavir) wird nicht empfohlen (siehe Abschnitt 4.4).
Arzneimittel, die den pH-Wert des Magens beeinflussen
Die Verabreichung von Ulipristalacetat (10 mg Tabletten) in Kombination mit dem Protonenpumpenhemmer Esomeprazol (20 mg täglich für 6 Tage) führte zu einer Verringerung der mittleren Cmax um etwa 65 %, einer Verzögerung der Tmax (von einem Median von 0,75 Stunden auf 1,0 .). Stunden) und einen Anstieg des Mittelwerts der AUC um 13 %. Diese Wirkung von Arzneimitteln, die den pH-Wert des Magens erhöhen, dürfte bei der täglichen Verabreichung von Ulipristalacetat in Tablettenform klinisch nicht relevant sein.
Mögliche Wechselwirkungen von Ulipristalacetat mit anderen Arzneimitteln:
Hormonelle Verhütungsmittel
Ulipristalacetat kann die Wirkung von hormonellen Kontrazeptiva (nur Gestagene, Gestagen freisetzende Geräte oder kombinierte orale Antibabypillen) und Gestagenen, die aus anderen Gründen verabreicht werden, beeinträchtigen. Die gleichzeitige Anwendung von Gestagen-haltigen Arzneimitteln wird daher nicht empfohlen (siehe Abschnitte 4.4 und 4.6). ) Arzneimittel, die Gestagene enthalten, sollten nach Beendigung der Behandlung mit Ulipristalacetat 12 Tage lang nicht eingenommen werden.
P-gp-Substrate
Daten in vitro weisen darauf hin, dass Ulipristalacetat in klinisch relevanten Konzentrationen an der Magen-Darm-Wand während der Resorption ein Inhibitor von P-gp sein kann.Die gleichzeitige Gabe von Ulipristalacetat und einem Substrat von P-gp wurde nicht untersucht und eine Wechselwirkung kann nicht ausgeschlossen werden. Die Ergebnisse in vivo zeigen, dass Ulipristalacetat (als Einzeltablette zu 10 mg) 1,5 Stunden vor der Verabreichung des P-gp-Substrats Fexofenadin (60 mg) keine klinisch relevante Wirkung auf die Pharmakokinetik von Fexofenadin hat. Es wird daher empfohlen, die gleichzeitige Anwendung von Ulipristalacetat und P-gp-Substraten (z. B. Dabigatranethexylat, Digoxin, Fexofenadin) um mindestens 1,5 Stunden zu verschieben.
04.6 Schwangerschaft und Stillzeit
Verhütung für Frauen
Es ist wahrscheinlich, dass Ulipristalacetat negativ mit reinen Gestagen-Pillen, Gestagen-freisetzenden Geräten oder kombinierten oralen Antibabypillen interagiert; Die gleichzeitige Anwendung wird daher nicht empfohlen.Obwohl die meisten Frauen, die eine therapeutische Dosis von Ulipristalacetat einnehmen, eine Anovulation aufweisen, wird die Anwendung einer nicht-hormonellen Verhütungsmethode während der Behandlung empfohlen (siehe Abschnitte 4.4 und 4.5).
Schwangerschaft
Ulipristalacetat ist während der Schwangerschaft kontraindiziert (siehe Abschnitt 4.3).
Es liegen keine oder nur begrenzte Daten zur Anwendung von Ulipristalacetat bei Schwangeren vor.
Obwohl kein teratogenes Potenzial festgestellt wurde, reichen die Daten zu Tierarten für eine Bewertung der Reproduktionstoxizität nicht aus (siehe Abschnitt 5.3).
Fütterungszeit
Die verfügbaren toxikologischen Daten von Tieren zeigten, dass Ulipristalacetat in die Muttermilch übergeht (Einzelheiten siehe Abschnitt 5.3). Ulipristalacetat geht in die Muttermilch über. Auswirkungen auf Säuglinge wurden nicht untersucht. Risiken für das Neugeborene können nicht ausgeschlossen werden. Ulipristalacetat ist während der Stillzeit kontraindiziert (siehe Abschnitte 4.3 und 5.2).
Fruchtbarkeit
Die Mehrheit der Frauen, die eine therapeutische Dosis von Ulipristalacetat einnehmen, weist eine Anovulation auf, jedoch wurde die Fertilität bei Einnahme mehrerer Dosen von Ulipristalacetat nicht untersucht.
04.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Ulipristalacetat kann die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen leicht beeinträchtigen, da nach Einnahme von Ulipristalacetat leichter Schwindel beobachtet wurde.
04.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Ulipristalacetat wurde bei 1.053 Frauen mit Uterusmyomen untersucht, die während Phase-III-Studien mit 5 mg oder 10 mg Ulipristalacetat behandelt wurden. Die häufigste Wirkung in klinischen Studien war Amenorrhoe (79,2 %), die als wünschenswertes Ergebnis für die Patienten angesehen wurde (siehe Abschnitt 4.4).
Die häufigste Nebenwirkung war Hitzewallung. Die überwiegende Mehrheit der Nebenwirkungen war leicht bis mittelschwer (95,0 %), führte nicht zum Absetzen des Arzneimittels (98,0 %) und klang spontan ab.
In dieser Gruppe von 1.053 Frauen wurde die Sicherheit wiederholter intermittierender Behandlungszyklen (jeweils auf 3 Monate begrenzt) bei 551 Frauen mit Uterusmyomen untersucht, die mit 5 oder 10 mg Ulipristalacetat in zwei Phase-III-Studien behandelt wurden (einschließlich 457 exponierten Frauen vier Kurse einer intermittierenden Behandlung), bei der das Arzneimittel ein ähnliches Sicherheitsprofil wie bei nur einer Behandlung aufwies.
Tabelle der Nebenwirkungen
Basierend auf gepoolten Daten aus vier Phase-III-Studien bei Patientinnen mit Uterusmyomen, die 3 Monate lang behandelt wurden, wurden die folgenden Nebenwirkungen berichtet. Die unten aufgeführten Nebenwirkungen sind nach Häufigkeit und Systemorgan klassifiziert. Innerhalb jeder Häufigkeitsklasse sind die Nebenwirkungen nach abnehmendem Schweregrad aufgelistet.Häufigkeiten sind definiert als sehr häufig (≥1/10), häufig (≥1/100 bis
* siehe Abschnitt „Beschreibung einiger Nebenwirkungen“
** der wörtliche Begriff „leichter Haarausfall“ wurde mit dem Begriff „Alopezie“ kodifiziert
Beim Vergleich wiederholter Behandlungszyklen war die Häufigkeit von Nebenwirkungen in den nachfolgenden Behandlungszyklen insgesamt geringer als im ersten Behandlungszyklus, und jede Nebenwirkung trat seltener auf oder blieb in der gleichen Häufigkeitskategorie (mit Ausnahme von Dyspepsie, die im Behandlungszyklus 3 als gelegentlich eingestuft wurde) wie es bei einem Patienten auftrat).
Beschreibung einiger Nebenwirkungen
Verdickung des Endometriums
Bei 10-15% der Patienten wurde unter Ulipristalacetat am Ende der ersten 3-monatigen Behandlung eine Verdickung des Endometriums (> 16 mm durch Ultraschall oder MRT am Ende der Behandlung) beobachtet. 4,9% bzw. 3,5% der Patientinnen am Ende des zweiten und vierten Behandlungszyklus) Die Verdickung des Endometriums verschwindet, wenn die Behandlung beendet und die Menstruationszyklen wieder aufgenommen werden.
Darüber hinaus werden die reversiblen Veränderungen des Endometriums als PAEC bezeichnet und unterscheiden sich von der Endometriumhyperplasie. Bei der Zusendung von Hysterektomie- oder Endometriumbiopsieproben zur histologischen Untersuchung sollte der Pathologe darüber informiert werden, dass der Patient Ulipristalacetat eingenommen hat (siehe Abschnitte 4.4 und 5.1).
Hitzewallungen
Hitzewallungen wurden von 8,1 % der Patienten berichtet, die Häufigkeit war jedoch von Studie zu Studie unterschiedlich.In der mit dem Wirkstoff kontrollierten Studie betrugen die Raten 24 % (10,5% mäßig oder schwer) mit Ulipristalacetat und 60,4 % (39,6 % mittel oder schwer) ) bei mit Leuprorelin behandelten Patienten.In der placebokontrollierten Studie betrug die Hitzewallung 1,0 % für Ulipristalacetat und 0 % für Placebo. Im ersten 3-monatigen Behandlungszyklus der beiden Langzeit-Phase-III-Studien betrug die Häufigkeit mit Ulipristalacetat 5,3 % bzw. 5,8 %.
Kopfschmerzen
Kopfschmerzen von leichter oder mittelschwerer Schwere wurden von 5,8 % der Patienten berichtet.
Eierstockzyste
Bei 1,0% der Patientinnen wurden während und nach der Behandlung funktionelle Ovarialzysten beobachtet, die in den meisten Fällen innerhalb weniger Wochen spontan verschwanden.
Gebärmutterblutung
Bei Patienten mit starken Menstruationsblutungen aufgrund von Gebärmuttermyomen besteht das Risiko einer übermäßigen Blutung, die eine Operation erforderlich machen kann.Einige Fälle wurden während der Behandlung mit Ulipristalacetat und innerhalb von 2-3 Monaten nach Beendigung der Behandlung mit Ulipristalacetat berichtet.
Meldung von vermuteten Nebenwirkungen
Die Meldung vermuteter Nebenwirkungen, die nach der Zulassung des Arzneimittels aufgetreten sind, ist wichtig, da sie eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht. Angehörige von Gesundheitsberufen werden gebeten, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem zu melden. in „Anhang V .
04.9 Überdosierung
Die Erfahrungen mit einer Überdosierung von Ulipristalacetat sind schlecht.
Einer begrenzten Anzahl von Probanden wurden an 10 aufeinanderfolgenden Tagen Einzeldosen des Arzneimittels bis zu 200 mg und Tagesdosen von 50 mg verabreicht, ohne dass schwerwiegende oder schwerwiegende Nebenwirkungen beobachtet wurden.
05.0 PHARMAKOLOGISCHE EIGENSCHAFTEN
05.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Sexualhormone und Modulatoren des Genitalsystems, Progesteronrezeptor-Modulatoren.
ATC-Code: G03XB02.
Ulipristalacetat ist ein oral wirksamer synthetischer selektiver Progesteronrezeptor-Modulator, der durch eine gewebespezifische partielle antagonistische Wirkung gegen Progesteron gekennzeichnet ist.
Endometrium
Ulipristalacetat hat eine direkte Wirkung auf die Gebärmutterschleimhaut.Wenn mit der täglichen Verabreichung einer Dosis von 5 mg während eines Menstruationszyklus begonnen wird, werden die meisten Menschen (einschließlich Patienten mit Myomen) ihre erste Periode beenden, aber die nächste Periode erst am Ende haben Behandlungsverlauf Nach Beendigung der Behandlung mit Ulipristalacetat werden die Menstruationszyklen in der Regel innerhalb von 4 Wochen wieder aufgenommen.
Die direkte Wirkung auf das Endometrium verursacht spezifische histologische Veränderungen dieser Kategorie von Arzneimitteln und definierter PAEC. Typischerweise ist der histologische Aspekt ein inaktives und schwach proliferierendes Epithel, das mit einer Asymmetrie des Stroma- und Epithelwachstums verbunden ist, das prominente Drüsen mit zystischer Dilatation und kombinierten Östrogenen ( mitotische) und Gestagen (sekretorische) epitheliale Wirkungen. Dieses Muster wurde bei etwa 60 % der Patienten beobachtet, die 3 Monate lang mit Ulipristalacetat behandelt wurden. Diese Veränderungen sind nach Beendigung der Behandlung reversibel. Diese Veränderungen sollten nicht mit einer Endometriumhyperplasie verwechselt werden.
Etwa 5 % der Patientinnen im gebärfähigen Alter mit starker Menstruation haben eine Endometriumdicke von mehr als 16 mm. Bei ca. 10-15% der mit Ulipristalacetat behandelten Patienten kann sich das Endometrium während der ersten 3-monatigen Behandlung verdicken (> 16 mm). Bei wiederholten Behandlungszyklen wurde eine geringe Häufigkeit der Endometriumverdickung festgestellt (4,9 % der Patienten nach dem zweiten Behandlungszyklus und 3,5 % nach dem vierten Behandlungszyklus). Die Verdickung verschwindet nach Absetzen der Behandlung und Wiederaufnahme der Menstruation. Für den Fall, dass die Endometriumverdickung nach Wiederaufnahme der Menstruation während der Behandlungsunterbrechung oder über die 3 Monate nach dem Ende der Behandlungszyklen hinaus fortbesteht, können weitere Untersuchungen gemäß der üblichen klinischen Praxis erforderlich sein, um andere zugrunde liegende Pathologien auszuschließen .
Myome
Ulipristalacetat übt eine direkte Wirkung auf Myome aus, die ihre Größe reduzieren, indem es die Zellproliferation hemmt und Apoptose induziert.
Hypophyse
Eine tägliche Dosis von 5 mg Ulipristalacetat hemmt bei den meisten Patientinnen den Eisprung, was durch konstante Progesteronspiegel um 0,3 ng/ml angezeigt wird.
Eine tägliche Dosis von 5 mg Ulipristalacetat unterdrückt die FSH-Spiegel teilweise, aber die Serumöstradiolspiegel werden bei den meisten Patienten im mittleren follikulären Bereich gehalten und ähneln den Spiegeln bei Patienten, die Placebo erhalten.
Ulipristalacetat hat keinen Einfluss auf die Serum-TSH-, ACTH- oder Prolaktinspiegel.
Klinische Wirksamkeit und Sicherheit
Präoperative Anwendung:
Die Wirksamkeit von festen Dosen von 5 mg und 10 mg Ulipristalacetat einmal täglich wurde in zwei 13-wöchigen randomisierten, doppelblinden Phase-III-Studien bei Patienten mit starker Menstruation im Zusammenhang mit Uterusmyomen untersucht Es wurde erwartet, dass die Patienten in dieser Studie zu Studienbeginn anämisch waren (Hb-Eisen oral, 80 mg Fe ++, plus Prüfpräparat. Studie 2 umfasste einen aktiven Vergleichspräparat, Leuprorelin 3,75 mg einmal monatlich mit einer intramuskulären Injektion Placebo wurde verwendet, um die verblindete Studie 2 aufrechtzuerhalten. In beiden Studien wurde der Menstruationsblutverlust anhand des Pictorial Bleeding Assessment Chart (PBAC) bewertet. Ein PBAC-Wert von > 100 in den ersten 8 Tagen der Menstruation wurde als Hinweis auf einen übermäßigen Menstruationsblutverlust angesehen.
In Studie 1 wurde ein statistisch signifikanter Unterschied in der Verringerung des menstruellen Blutverlusts zugunsten von mit Ulipristalacetat behandelten Patienten im Vergleich zu Placebo beobachtet (siehe Tabelle 1 unten), was zu einer schnelleren und effizienteren Korrektur der Anämie führte Eisen allein. In ähnlicher Weise zeigten Patienten, die mit Ulipristalacetat behandelt wurden, eine stärkere Verringerung der Myome in der MRT.
In Studie 2 war die Verringerung des menstruellen Blutverlustes bei Patienten, die mit Ulipristalacetat und dem Gonadotropin-Releasing-Hormon (Leuprorelin)-Agonisten behandelt wurden, ähnlich. Die meisten mit Ulipristalacetat behandelten Patienten hörten innerhalb der ersten Behandlungswoche auf (Amenorrhoe).
Die Größe der drei größten Myome wurde am Ende der Behandlung (Woche 13) und für weitere 25 Wochen ohne Behandlung bei Patienten ohne Hysterektomie oder Myomektomie mit Ultraschall beurteilt.Die Verringerung der Myomgröße wurde dabei im Allgemeinen beibehalten bei Patienten, die mit Ulipristalacetat behandelt wurden, während bei Patienten, die mit Leuprorelin behandelt wurden, ein gewisses Nachwachsen auftrat.
Tabelle 1: Ergebnisse der primären Wirksamkeitsbewertungen und einiger sekundärer Wirksamkeitsbewertungen in Phase-III-Studien
a In Studie 1 wurde die Veränderung des Gesamtmyomvolumens gegenüber dem Ausgangswert mittels MRT gemessen. In Studie 2 wurde die Volumenänderung der drei größten Myome mit Ultraschall gemessen. Fettgedruckte Werte in den schattierten Kästchen weisen auf einen signifikanten Unterschied im Vergleich zwischen Ulipristalacetat und der Kontrolle hin. Diese Unterschiede waren immer zugunsten von Ulipristalacetat.
P-Werte: 1 =
Intermittierender wiederholter Gebrauch:
Die Wirksamkeit wiederholter Behandlungszyklen mit festen Dosen von Ulipristalacetat 5 mg oder 10 mg einmal täglich wurde in zwei Phase-3-Studien untersucht, in denen bis zu 4 intermittierende 3-monatige Behandlungszyklen bei Patienten mit sehr starker Menstruation im Zusammenhang mit Uterusmyomen analysiert wurden eine offene Studie zur Untersuchung von Ulipristalacetat 10 mg, in der auf jede 3-monatige Behandlung eine 10-tägige doppelblinde Gestagen- oder Placebo-Behandlung folgte.Studie 4 war eine randomisierte klinische Studie, doppelblinde Studie, bei der Ulipristalacetat 5 oder 10 mg.
Die Studien 3 und 4 zeigten die Wirksamkeit bei der Kontrolle der Symptome von Uterusmyomen (z. B. Uterusblutungen) und bei der Verringerung der Größe des Myoms nach 2 und 4 Behandlungszyklen.
In Studie 3 wurde die Wirksamkeit der Behandlung bei wiederholter intermittierender Behandlung über einen Zeitraum von mehr als 18 Monaten (4 Zyklen mit 10 mg einmal täglich) nachgewiesen; 89,7 % der Patienten hatten am Ende der Behandlung eine Amenorrhoe 4.
In Studie 4 hatten 61,9% und 72,7% der Patienten Amenorrhoe am Ende der Behandlungszyklen 1 und 2 kombiniert (5 mg Dosis bzw. 10 mg Dosis, p = 0,032); 48,7% und 60,5% der Patienten hatten Amenorrhoe am Ende aller vier Behandlungszyklen (5 mg Dosis bzw. 10 mg Dosis, p = 0,027). Am Ende von Behandlungszyklus 4 hatten 158 Patienten (69,6 %) und 164 Patienten (74,5 %) eine Amenorrhoe bei der 5-mg- bzw. 10 mg-Dosis (p = 0,290).
Tabelle 2: Ergebnisse der primären Wirksamkeitsbewertungen und einiger sekundärer Wirksamkeitsbewertungen in langfristigen Phase-III-Studien
a Die Beurteilung des Behandlungszyklus 2 entspricht dem Behandlungszyklus 2 plus einer Menstruationsblutung.
b Patienten, für die Daten fehlten, wurden von der Analyse ausgeschlossen.
c N und % beinhalten zurückgezogene Patienten
Kontrollierte Blutungen wurden definiert als keine Episoden starker Blutungen und maximal 8 Tage Blutung (ausgenommen Schmiertage) während der letzten zwei Monate eines Behandlungszyklus.
Endometriumbefunde:
In allen Phase-III-Studien, einschließlich intermittierender Wiederholungsbehandlungsstudien, wurden bei 789 Patienten mit adäquaten Biopsien insgesamt 7 Fälle von Hyperplasie beobachtet (0,89%). In den allermeisten Fällen normalisierte sich das Endometrium nach Wiederaufnahme der Menstruation während der Behandlungsabsetzzeit spontan wieder.Die Inzidenz von Hyperplasie nahm bei wiederholten Behandlungszyklen nicht zu. Die beobachtete Häufigkeit entspricht der der Kontrollgruppen und der in der Literatur berichteten Prävalenz für symptomatische prämenopausale Frauen dieser Altersgruppe (Mittelwert 40 Jahre).
Die Europäische Arzneimittel-Agentur hat auf die Verpflichtung zur Vorlage der Ergebnisse von Studien mit Esmya in allen Untergruppen der pädiatrischen Population bei Uterusmyomen verzichtet (siehe Abschnitt 4.2 für Informationen zur pädiatrischen Anwendung).
05.2 Pharmakokinetische Eigenschaften
Absorption
Nach oraler Verabreichung einer Einzeldosis von 5 oder 10 mg wird Ulipristalacetat schnell resorbiert, mit einer Cmax von 23,5 ± 14,2 ng/ml und 50,0 ± 34,4 ng/ml etwa 1 Stunde nach der Einnahme und einer AUC0-∞ von 61,3 ± 31,7 ng.h/ml bzw. 134,0 ± 83,8 ng.h/ml Ulipristalacetat wird rasch in einen pharmakologisch aktiven Metaboliten mit Cmax von 9,0 ± 4,4 ng/ml und 20,6 ± 10,9 ng/ml umgewandelt, wiederum etwa 1 Stunde nach Einnahme und AUC0-∞ von 26,0 ± 12,0 ng.h/ml bzw. 63, 6 ± 30,1 ng.h/ml.
Die Einnahme von Ulipristalacetat (30 mg Tablette) zusammen mit einem fettreichen Frühstück führte zu einer Verringerung der mittleren Cmax um ca. 45 %, einer Verzögerung der Tmax (Median 0,75 bis 3 Stunden) und einer 25 %igen Erhöhung des mittleren AUC0-∞-Wertes im Vergleich zur Dosierung im nüchternen Zustand. Ähnliche Ergebnisse wurden für den aktiven Mono-N-desmethyl-Metaboliten erhalten. Es wird nicht erwartet, dass diese kinetische Wirkung von Nahrungsmitteln bei der täglichen Verabreichung von Ulipristalacetat-Tabletten klinisch relevant ist.
Verteilung
Ulipristalacetat bindet stark (> 98 %) an Plasmaproteine, einschließlich Albumin, alpha-1-saures Glykoprotein, High-Density-Lipoprotein und Low-Density-Lipoprotein.
Ulipristalacetat und sein aktiver mono-N-demethylierter Metabolit gehen mit einem Milch/Plasma-AUCt-Verhältnis von 0,74 ± 0,32 für Ulipristalacetat in die Muttermilch über.
Biotransformation / Elimination
Ulipristalacetat wird schnell in seine mono-N-demethylierten und anschließend in seine di-N-demethylierten Metaboliten umgewandelt. Die Daten in vitroweisen darauf hin, dass diese Umwandlung überwiegend durch die Cytochrom-P450-Isoform 3A4 (CYP3A4) vermittelt wird. Der primäre Eliminationsweg erfolgt über die Fäzes und weniger als 10 % werden mit dem Urin ausgeschieden. Die geschätzte terminale Halbwertszeit von Ulipristalacetat im Plasma nach einmaligem Die Dosis von 5 oder 10 mg wird auf ungefähr 38 Stunden geschätzt, mit einer mittleren oralen Clearance (CL/F) von ungefähr 100 l/h.
Die Daten in vitro weisen darauf hin, dass Ulipristalacetat und sein aktiver Metabolit weder CYP1A2, 2A6, 2C9, 2C19, 2D6, 2E1 und 3A4 hemmen noch CYP1A2 in klinisch relevanten Konzentrationen induzieren. Daher ist es unwahrscheinlich, dass Ulipristalacetat die Clearance von Arzneimitteln verändert, die durch diese Enzyme metabolisiert werden.
Die Daten in vitro weisen darauf hin, dass Ulipristalacetat und sein aktiver Metabolit keine Substrate von P-gp (ABCB1) sind.
Besondere Bevölkerungsgruppen
Bei Frauen mit eingeschränkter Nieren- oder Leberfunktion wurden keine pharmakokinetischen Studien mit Ulipristalacetat durchgeführt. Aufgrund der CYP-vermittelten Metabolisierung ist zu erwarten, dass eine Leberinsuffizienz die Elimination von Ulipristalacetat verändert, was zu einer erhöhten Exposition führt (siehe Abschnitte 4.2 und 4.4).
05.3 Präklinische Sicherheitsdaten
Präklinische Daten lassen auf der Grundlage konventioneller Studien zu keine besonderen Gefahren für den Menschen erkennen Sicherheitspharmakologie, Toxizität bei wiederholter Gabe und Genotoxizität.
Die meisten Ergebnisse aus allgemeinen Toxizitätsstudien bezogen sich auf die Wirkung auf Progesteron-Rezeptoren (und höhere Konzentrationen auf Glucocorticoid-Rezeptoren) und zeigten eine Antiprogesteron-Wirkung bei Expositionen ähnlich den therapeutischen Konzentrationen.In einer 39-wöchigen Studie an Cynomolgus-Affen waren PAEC-ähnliche histologische Veränderungen bei niedrigen Dosen bemerkt.
Aufgrund seines Wirkmechanismus hat Ulipristalacetat eine embryoletale Wirkung bei Ratten, Kaninchen (bei wiederholten Dosen über 1 mg / kg), Meerschweinchen und Affen Es liegen keine Daten zur Sicherheit des menschlichen Embryos vor. Bei Dosen, die niedrig genug waren, um die Trächtigkeit bei Tierarten aufrechtzuerhalten, wurde kein teratogenes Potenzial beobachtet.
Reproduktionsstudien, die an Ratten mit Dosen durchgeführt wurden, die eine Exposition ähnlich der beim Menschen verwendeten Dosis bewirkten, zeigten keine Hinweise auf eine Schädigung der Fertilität durch Ulipristalacetat bei behandelten Tieren oder bei den Nachkommen behandelter weiblicher Tiere.
Kanzerogenitätsstudien (an Ratten und Mäusen) zeigten, dass Ulipristalacetat nicht krebserregend ist.
06.0 PHARMAZEUTISCHE INFORMATIONEN
06.1 Hilfsstoffe
Mikrokristalline Cellulose
Mannit
Croscarmellose-Natrium
Talk
Magnesiumstearat
06.2 Inkompatibilität
Nicht relevant.
06.3 Gültigkeitsdauer
3 Jahre.
06.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Bewahren Sie die Blisterpackungen im Umkarton auf, um das Arzneimittel vor Licht zu schützen.
06.5 Art der unmittelbaren Verpackung und Inhalt des Packstücks
Al / PVC / PE / PVDC oder Al / PVC / PVDC Blister.
Packungen mit 28, 30 und 84 Tabletten.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
06.6 Gebrauchs- und Handhabungshinweise
Keine besonderen Anweisungen.
07.0 INHABER DER MARKETING-ERLAUBNIS
Gedeon Richter Plc.
Gyömroi út 19-21.
1103 Budapest
Ungarn
08.0 NUMMER DER MARKETING-ERLAUBNIS
EU / 12.01.750/001
042227013
EU / 12.01.750/002
042227025
EU / 12.01.750/003
042227037
EU / 12.01.750/004
042227049
EU / 12.01.750/005
042227052
09.0 DATUM DER ERSTEN GENEHMIGUNG ODER ERNEUERUNG DER GENEHMIGUNG
Datum der Erstzulassung: 23. Februar 2012
10.0 DATUM DER ÜBERARBEITUNG DES TEXTs
D.CCE Mai 2015