Wirkstoffe: Epoetin zeta
Retacrit 1 000 IE / 0,3 ml Injektionslösung in einer Fertigspritze
Retacrit 2 000 IE / 0,6 ml Injektionslösung in einer Fertigspritze
Retacrit 3.000 IE / 0,9 ml Injektionslösung in einer Fertigspritze
Retacrit 4 000 IE / 0,4 ml Injektionslösung in einer Fertigspritze
Retacrit 5 000 IE / 0,5 ml Injektionslösung in einer Fertigspritze
Retacrit 6 000 IE / 0,6 ml Injektionslösung in einer Fertigspritze
Retacrit 8 000 IE / 0,8 ml Injektionslösung in einer Fertigspritze
Retacrit 10 000 IE / 1 ml Injektionslösung in einer Fertigspritze
Retacrit 20 000 IE / 0,5 ml Injektionslösung in einer Fertigspritze
Retacrit 30 000 IE / 0,75 ml Injektionslösung in einer Fertigspritze
Retacrit 40 000 IE / 1 ml Injektionslösung in einer Fertigspritze
Indikationen Warum wird Retacrit angewendet? Wofür ist das?
Retacrit enthält ein Protein namens Epoetin zeta, das das Knochenmark anregt, mehr rote Blutkörperchen im Blut zu bilden, die Hämoglobin (eine Substanz, die Sauerstoff bindet) tragen. Epoetin zeta ist eine Kopie des menschlichen Proteins Erythropoietin und wirkt auf die gleiche Weise.
Retacrit wird verwendet:
- bei erwachsenen, pädiatrischen und jugendlichen Patienten, die sich einer Hämodialyse unterziehen, zur Behandlung von symptomatischer Anämie (verminderte Anzahl roter Blutkörperchen) in Verbindung mit chronischem Nierenversagen (Nierenerkrankung);
- bei erwachsenen Patienten, die sich einer Peritonealdialyse unterziehen, zur Behandlung von symptomatischer Anämie im Zusammenhang mit chronischem Nierenversagen (Nierenerkrankung);
- bei erwachsenen Patienten mit Niereninsuffizienz, die sich noch nicht einer Dialyse unterziehen, zur Behandlung einer schweren Anämie in Verbindung mit einer Nierenerkrankung, die von klinischen Symptomen begleitet wird;
- bei erwachsenen Patienten, die eine Chemotherapie bei soliden Tumoren, malignen Lymphomen (Krebs des Lymphsystems) oder multiplem Myelom (Knochenmarkkrebs) erhalten, um eine Anämie zu behandeln und den Bedarf an Bluttransfusionen zu verringern, wenn der Arzt ein hohes Risiko für Transfusionen benötigen; - bei Patienten mit mittelschwerer Anämie, die für eine Operation in Frage kommen, um vor der Operation Blut zu spenden, damit sie während oder nach der Operation eigenes Blut erhalten können (Eigenblutspende);
- bei erwachsenen Patienten mit mäßiger Anämie, bei denen größere orthopädische (Knochen-)Operationen (z. B. Hüft- oder Knieersatztherapie) geplant sind, um den Bedarf an Bluttransfusionen zu verringern.
Kontraindikationen Wenn Retacrit nicht angewendet werden sollte
Verwenden Sie Retacrit nicht:
- wenn Sie allergisch gegen Erythropoietin oder einen der in Abschnitt 6. genannten sonstigen Bestandteile dieses Arzneimittels sind.
- wenn Sie nach einer Behandlung mit einer beliebigen Art von Erythropoietin an einer Krankheit namens „Reine Red Cell Aplasie“ (PRCA) erkrankt sind
- wenn Sie hohen Blutdruck haben, der mit bestimmten blutdrucksenkenden Arzneimitteln nicht ausreichend kontrolliert werden kann
- wenn Sie keine Arzneimittel zur Blutverdünnung einnehmen können
- wenn Sie vor einer Operation Blut spenden und:
- im Monat vor der Behandlung einen Herzinfarkt oder Schlaganfall hatten
- an instabiler Angina pectoris leiden (neue oder zunehmende Brustschmerzen)
- die Gefahr der Bildung von Blutgerinnseln in den Venen besteht (tiefe Venenthrombose); wenn Sie beispielsweise schon einmal an einer Thrombose gelitten haben.
- Wenn bei Ihnen ein größerer orthopädischer Eingriff wie Hüft- oder Kniegelenkersatz geplant ist und:
- wenn Sie schwere Herz- oder Durchblutungsstörungen in Venen oder Arterien haben
- wenn Sie kürzlich einen Herzinfarkt oder Schlaganfall hatten.
Vorsichtsmaßnahmen für die Anwendung Was sollten Sie vor der Einnahme von Retacrit® beachten?
Informieren Sie vor der Anwendung von Retacrit Ihren Arzt, wenn Sie wissen, dass Sie an einer der folgenden Krankheiten gelitten haben oder leiden:
- Anfälle
- Leber erkrankung
- Tumore
- Anämie aufgrund anderer Ursachen
- Herzerkrankungen (wie Angina pectoris)
- Durchblutungsstörungen, die ein Kribbeln in den Extremitäten, kalte Hände oder Füße oder Muskelkrämpfe in den Beinen verursachen
- Thrombose oder Gerinnungskrankheiten
- Nierenerkrankung.
Wechselwirkungen Welche Medikamente oder Lebensmittel können die Wirkung von Retacrit® verändern?
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen, kürzlich andere Arzneimittel eingenommen haben oder beabsichtigen andere Arzneimittel einzunehmen.
Insbesondere wenn Sie zur Hemmung des Immunsystems nach einer Nierentransplantation ein Arzneimittel einnehmen, das den Wirkstoff Ciclosporin enthält, kann Ihr Arzt während der Behandlung mit Retacrit spezielle Tests zur Messung der Ciclosporinkonzentration im Blut anordnen.
Die Einnahme von Eisenpräparaten und anderen Blutstimulanzien kann die Wirksamkeit von Retacrit erhöhen. Ihr Arzt wird entscheiden, ob Sie diese Substanzen weiterhin einnehmen sollen.
Warnungen Es ist wichtig zu wissen, dass:
Während der Behandlung mit Retacrit®
Ihr Arzt wird überprüfen, ob Ihr Hämoglobinwert einen bestimmten Wert nicht überschreitet, da hohe Hämoglobinkonzentrationen die Gesundheit des Herzens oder der Blutgefäße gefährden und das Risiko für Herzinfarkt, Schlaganfall und Tod erhöhen können.
Ärzte sollten versuchen, den Hämoglobinspiegel zwischen 10 und 12 g/dl zu halten. Der Hämoglobinspiegel sollte 12 g / dl nicht überschreiten.
Ihr Arzt wird Ihren Blutdruck während der Anwendung von Retacrit regelmäßig überprüfen.Wenn Sie Kopfschmerzen, insbesondere plötzliche, pochende Migräne, verspüren oder wenn Sie sich verwirrt fühlen oder Anfälle haben, informieren Sie sofort Ihren Arzt oder das medizinische Fachpersonal.
Diese Symptome könnten in der Tat Warnzeichen für einen plötzlichen Blutdruckanstieg sein, der eine Notfallbehandlung erfordern würde.
Während der Behandlung mit diesem Arzneimittel kann ein Anstieg der Blutplättchen (Zellen, die zur Blutgerinnung beitragen) auftreten. Dieses Phänomen sollte sich im Laufe der Behandlung bessern. Wir empfehlen Ihnen, Ihre Thrombozytenzahl während der ersten 8 Wochen der Therapie regelmäßig zu überprüfen.
Wenn Sie sich einer medizinischen Untersuchung in einem Krankenhaus oder einer Privatklinik unterziehen oder sich Blutuntersuchungen unterziehen, denken Sie daran, Ihren Arzt über die von Ihnen durchgeführte Retacrit-Behandlung zu informieren, da dieses Arzneimittel Ihren Zustand und Ihre Testergebnisse verändern kann.
Achten Sie besonders auf andere Produkte, die die Produktion von roten Blutkörperchen anregen:
Retacrit gehört ebenso wie das menschliche Protein Erythropoietin zu einer Gruppe von Produkten, die die Produktion von roten Blutkörperchen stimulieren. Das medizinische Fachpersonal wird immer den genauen Namen des von ihm verwendeten Produkts aufzeichnen.
Patienten mit Nierenerkrankungen
In seltenen Fällen wurde nach monatelanger oder jahrelanger Behandlung mit anderen Erythropoietin-haltigen Arzneimitteln über reine Erythrozyten-Aplasie (PRCA) berichtet; diese Möglichkeit kann bei Retacrit nicht ausgeschlossen werden.
Spezifische Aplasie der roten Blutkörperchen beinhaltet die Unfähigkeit des Knochenmarks, genügend rote Blutkörperchen zu produzieren. In diesem Fall kann eine schwere Anämie auftreten, deren Symptome sind: ungewöhnliche Müdigkeit, Schwindel oder Kurzatmigkeit. Die Erythropoietin-Aplasie kann durch die Produktion von Antikörpern verursacht werden, die gegen das injizierte Erythropoietin und anschließend gegen das vom gleichen Organismus produzierte Erythropoietin gerichtet sind.
Besprechen Sie diese Informationen mit Ihrem Arzt. Wenn diese Aplasie, wie auch immer selten, auftritt, wird die Behandlung mit Retacrit ausgesetzt und der Arzt wird entscheiden, wie die Anämie am effektivsten behandelt werden kann. Sie müssen die Einnahme von Retacrit abbrechen und regelmäßig und möglicherweise lebenslang Bluttransfusionen zur Behandlung von Anämie erhalten. Informieren Sie unverzüglich Ihren Arzt, wenn Sie sich plötzlich sehr müde oder kurzatmig fühlen.Ihr Arzt wird feststellen, wie wirksam Retacrit bei Ihnen ist, und die Behandlung gegebenenfalls abbrechen.
Patienten mit chronischer Niereninsuffizienz, die mit Erythropoietin behandelt werden, müssen in regelmäßigen Abständen Tests unterzogen werden, um den Hämoglobinspiegel (der Teil der roten Blutkörperchen, der Sauerstoff transportiert) zu messen, bis ein konstanter Spiegel erreicht ist Gefahr eines Blutdruckanstiegs.
Wenn Sie an chronischer Niereninsuffizienz leiden und insbesondere nicht ausreichend auf Retacrit ansprechen, wird Ihr Arzt die Retacrit-Dosis, die Sie erhalten, überprüfen, denn wenn Sie auf die Behandlung nicht ansprechen, kann eine wiederholte Erhöhung der Retacrit-Dosis das Risiko für Herz- oder Gefäßerkrankungen und kann das Risiko von Herzinfarkt, Schlaganfall und Tod erhöhen.
In Einzelfällen wurde ein Anstieg des Kaliumspiegels im Blut beobachtet. Bei Patienten mit chronischer Niereninsuffizienz kann die Korrektur der Anämie zu einer Zunahme des Appetits und der Aufnahme von Kalium und Protein führen.Wenn Sie sich zu Beginn der Behandlung mit Retacrit in einer Dialysebehandlung befinden, müssen Sie möglicherweise die Dialyseparameter anpassen, um Harnstoff, Kreatinin- und Kaliumspiegel innerhalb des gewünschten Bereichs und Ihr Arzt wird dies entscheiden.
Bei Patienten mit chronischer Niereninsuffizienz sollten die Serumelektrolyte (im Blut gefundene Substanzen) überwacht werden. Wenn die Serumkaliumwerte hoch (oder steigend) sind, sollte erwogen werden, die Verabreichung von Retacrit abzubrechen, bis diese Werte korrigiert sind.
Während der Therapie mit Retacrit ist es während der Hämodialyse häufig erforderlich, die Dosis von Heparin, einem bestimmten blutverdünnenden Stoff, zu erhöhen, um das Risiko von Blutgerinnseln zu minimieren.Wenn diese Dosis von Heparin nicht optimal ist, kann es zu einem Verschluss kommen der Dialysator.
Krebspatienten
Bei Krebspatienten ist die Wahrscheinlichkeit einer Thrombose höher, wenn sie Erythropoietin-ähnliche Arzneimittel wie Retacrit einnehmen (siehe Abschnitt 4) Sie sollten daher mit Ihrem Arzt über die Vorteile von Retacrit sprechen, insbesondere wenn Sie übergewichtig sind oder früher an Thromboseproblemen gelitten haben oder Blutgerinnungskrankheiten.
Krebspatienten, die mit Erythropoietin behandelt werden, müssen in regelmäßigen Abständen Labortests unterzogen werden, um den Hämoglobinspiegel (der Teil der roten Blutkörperchen, der Sauerstoff transportiert) zu messen, bis ein konstanter Spiegel erreicht ist, und danach mit Fristen.
Wenn Sie Krebs haben, sollten Sie sich bewusst sein, dass Retacrit als Wachstumsfaktor für Blutzellen wirken kann und unter Umständen negative Auswirkungen auf den Krebs haben kann. Je nach Situation kann eine Bluttransfusion vorzuziehen sein. Besprechen Sie dies mit Ihrem Arzt.
Schwangerschaft und Stillzeit
Wenn Sie schwanger sind oder stillen oder vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, fragen Sie vor der Einnahme dieses Arzneimittels Ihren Arzt oder Apotheker um Rat.
Wenn Sie schwanger sind oder stillen, sollte Retacrit nur angewendet werden, wenn der potenzielle Nutzen die potenziellen Risiken für das Baby überwiegt.
Fragen Sie Ihren Arzt um Rat, bevor Sie ein Arzneimittel einnehmen.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Retacrit hat keinen oder einen sehr geringen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Retacrit enthält Phenylalanin
Dieses Arzneimittel enthält Phenylalanin, eine Substanz, die für Menschen mit Phenylketonurie (ein Enzymmangel genetischen Ursprungs, der zu einer erhöhten Ausscheidung einer chemischen Substanz (Phenylketon) im Urin führt und Störungen des Nervensystems verursachen kann) gefährlich sein kann.
Retacrit enthält Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, was bedeutet, dass es als „natriumfrei“ gilt.
Dosierung und Art der Anwendung Wie ist Retacrit anzuwenden: Dosierung
Die Retacrit-Therapie wird in der Regel unter ärztlicher Aufsicht begonnen. Retacrit-Injektionen können von einem Arzt, einer Krankenschwester oder einem anderen medizinischen Fachpersonal verabreicht werden.
Wenn Retacrit unter die Haut (subkutan) injiziert wird, können Sie die Lösung auch selbst injizieren, nachdem Sie gesehen haben, wie es geht. Wenden Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt an. Wenn Sie sich nicht sicher sind, sprechen Sie mit Ihrem Arzt.
Dosisinformationen
Die Dosierung richtet sich nach dem Körpergewicht in Kilogramm. Ihr Arzt wird Tests, z. B. Bluttests, anordnen, um festzustellen, ob Sie Retacrit einnehmen müssen, und er wird die genaue Dosis von Retacrit bestimmen, wie lange Sie sich der Behandlung unterziehen müssen und wie das Arzneimittel verabreicht wird. Diese Entscheidungen hängen von der Ursache der Anämie ab.Ihr Arzt wird die niedrigste wirksame Dosis verwenden, um die Symptome der Anämie zu kontrollieren. Wenn Sie nicht ausreichend auf Retacrit ansprechen, wird Ihr Arzt die Dosis, die Sie erhalten, überprüfen und Sie informieren, wenn er sie ändert.
Sie können sowohl vor als auch während der Behandlung mit Retacrit Eisenpräparate erhalten, um die Wirksamkeit der Therapie zu erhöhen.
Anwendung bei Patienten mit Nierenerkrankungen
Retacrit muss entweder unter die Haut (subkutan) oder durch Injektion in eine Vene oder über einen in eine Vene gelegten Katheter verabreicht werden.
Anwendung von Retacrit bei erwachsenen Patienten, die Hämodialyse erhalten
Der Arzt wird die Hämoglobinkonzentration zwischen 10 und 12 g/dl (6,2 - 7,5 mmol/l) halten.
Retacrit kann während der Dialysesitzung oder am Ende der Sitzung verabreicht werden.
Die empfohlene Anfangsdosis beträgt 50 IE / kg (Internationale Einheiten pro Kilogramm) dreimal pro Woche. Wenn die Lösung in eine Vene verabreicht wird, sollte sie über 1 bis 5 Minuten injiziert werden.
Je nachdem, wie Ihre Anämie auf die Behandlung anspricht, kann diese Dosierung ungefähr alle 4 Wochen angepasst werden, bis die Situation unter Kontrolle ist.Ihr Arzt wird regelmäßige Blutuntersuchungen verschreiben, um sicherzustellen, dass das Arzneimittel weiterhin die gewünschte Wirkung hat. Sobald die Situation unter Kontrolle ist, werden Sie Retacrit weiterhin 2 oder 3 Mal pro Woche in regulären Dosen einnehmen. Diese Dosierungen sind möglicherweise nicht so hoch wie die ursprünglich erhaltenen.
Anwendung von Retacrit bei Kindern und Jugendlichen
(≤18 Jahre) mit Hämodialyse behandelt Bei Kindern wird der Arzt die Hämoglobinkonzentration zwischen 9,5 und 11 g/dl halten.
Retacrit sollte dem Patienten am Ende der Dialysesitzung verabreicht werden.
Die Dosierung für Kinder und Jugendliche richtet sich nach dem Körpergewicht in Kilogramm. Die empfohlene Anfangsdosis beträgt 50 IE / kg, verabreicht dreimal pro Woche durch Injektion in eine Vene (über eine Dauer von 1-5 Minuten).
Je nachdem, wie die Anämie auf die Behandlung anspricht, kann diese Dosis ungefähr alle 4 Wochen angepasst werden, bis die Situation unter Kontrolle ist.Ihr Arzt wird regelmäßige Blutuntersuchungen anordnen, um dies sicherzustellen.
Anwendung von Retacrit bei erwachsenen Patienten, die sich einer Peritonealdialyse unterziehen
Der Arzt wird die Hämoglobinkonzentration zwischen 10 und 12 g/dl halten.
Die empfohlene Anfangsdosis beträgt 50 IE / kg und wird zweimal wöchentlich verabreicht.
Je nachdem, wie die Anämie auf die Behandlung anspricht, kann diese Dosierung etwa alle 4 Wochen angepasst werden, bis die Situation unter Kontrolle ist.
Ihr Arzt wird Ihnen regelmäßig Blutuntersuchungen anordnen, um sicherzustellen, dass das Arzneimittel weiterhin die gewünschte Wirkung hat.
Anwendung von Retacrit bei erwachsenen Patienten mit Nierenerkrankung, die sich jedoch keiner Dialyse unterziehen
Die empfohlene Anfangsdosis beträgt 50 IE / kg, dreimal pro Woche verabreicht.
Diese Anfangsdosis kann von Ihrem Arzt angepasst werden, bis die Situation unter Kontrolle ist. Sobald die Situation unter Kontrolle ist, nehmen Sie Retacrit weiterhin in regelmäßigen Dosen (3-mal wöchentlich oder bei subkutaner Anwendung unter die Haut) auch einmal wöchentlich oder alle 2 Wochen ein 3-mal wöchentlich 150 IE / kg, einmal wöchentlich 240 IE / kg (bis maximal 20.000 IE) oder einmal wöchentlich 480 IE / kg (bis maximal 40.000 IE) überschreiten.
Ihr Arzt wird Ihnen regelmäßig Blutuntersuchungen anordnen, um sicherzustellen, dass das Arzneimittel weiterhin die gewünschte Wirkung hat.
Wenn Sie in längeren Dosierungsintervallen (mehr als einmal pro Woche) behandelt werden, können Sie Ihren Hb-Spiegel möglicherweise nicht ausreichend halten und müssen möglicherweise Ihre Retacrit-Dosis oder die Häufigkeit der Anwendung erhöhen.
Anwendung von Retacrit bei erwachsenen Patienten, die eine Chemotherapie erhalten
Ihr Arzt kann mit Retacrit beginnen, wenn Ihr Hämoglobinspiegel 10 g/dl oder weniger beträgt.
Nach Therapiebeginn wird der Arzt die Hämoglobinkonzentration zwischen 10 und 12 g/dl halten.
Die empfohlene Anfangsdosis beträgt 150 IE / kg und wird dreimal pro Woche subkutan injiziert. Alternativ kann Ihr Arzt eine Anfangsdosis von 450 IE / kg einmal pro Woche empfehlen. Ihr Arzt kann Ihre Anfangsdosis basierend auf Ihrem Ansprechen auf die Anämie auf die Behandlung anpassen; Sie werden Retacrit noch 1 Monat nach dem Ende der Chemotherapie einnehmen.
Anwendung bei erwachsenen Patienten, die an einem Programm zur Eigenblutspende teilnehmen
Die empfohlene Anfangsdosis beträgt 600 IE / kg, zweimal wöchentlich als Injektion in eine Vene. Retacrit wird Ihnen in den 3 Wochen vor der Operation verabreicht.Sie werden auch Eisenpräparate vor und während der Behandlung mit Retacrit einnehmen, um die Wirksamkeit dieses Arzneimittels zu erhöhen.
Anwendung bei erwachsenen Patienten, bei denen größere orthopädische (Knochen-)Operationen geplant sind
Eine Dosis von 600 I.E./kg wird einmal wöchentlich für 3 Wochen vor der Operation und am Tag der Operation unter die Haut gespritzt. In Fällen, in denen die Zeit vor dem Eingriff verkürzt werden muss, wird in den zehn Tagen vor dem Eingriff, am Tag des Eingriffs und in den 4 folgenden Tagen eine Dosis von 300 IE / kg verabreicht.Wenn Bluttests vor der Operation einen zu hohen Hämoglobinspiegel zeigen, wird die Behandlung abgebrochen.
Es ist auch wichtig, dass die Eisenwerte im Blut für die Dauer der Behandlung mit Retacrit normal sind. Bei Bedarf erhalten Sie täglich Eisen oral, vorzugsweise bereits vor Beginn der Behandlung mit Retacrit.
Informationen zur Verwaltung
Die Retacrit-Fertigspritze ist gebrauchsfertig. Jede Spritze ist nur zur einmaligen Injektion bestimmt. Retacrit-Injektionslösung darf nicht geschüttelt oder mit anderen Lösungen gemischt werden.
Wenn Retacrit unter die Haut injiziert wird, sollte die in eine einzelne Injektionsstelle injizierte Menge 1 ml nicht überschreiten. Der Oberschenkel und der Bauch weg vom Nabel sind gute Injektionsstellen. Wechseln Sie die Injektionsstelle täglich.
Befolgen Sie bei der Verwendung von Retacrit immer diese Anweisungen:
- Nehmen Sie die versiegelte Blisterpackung mit der Spritze und lassen Sie sie Raumtemperatur annehmen, bevor Sie sie verwenden. Dafür dauert es 15 bis 30 Minuten.
- Nehmen Sie die Spritze aus der Blisterpackung und prüfen Sie, ob die Lösung klar, farblos und praktisch frei von sichtbaren Partikeln ist.
- Entfernen Sie die Nadelabdeckung und lassen Sie die Luft aus der Nadel und Spritze, indem Sie die Spritze aufrecht halten und den Kolben vorsichtig nach oben drücken.
- Injizieren Sie die Lösung gemäß den Anweisungen Ihres Arztes. Wenn Ihnen etwas nicht klar ist, fragen Sie Ihren Arzt oder Apotheker.
Verwenden Sie Retacrit nicht, wenn:
- der Blister ist offen oder anderweitig beschädigt;
- die Lösung ist nicht farblos oder enthält sichtbare Schwebeteilchen;
- c „Flüssigkeit ist aus der Fertigspritze ausgetreten oder es ist Kondenswasser in der noch versiegelten Blisterpackung sichtbar;
- wenn Sie wissen, dass das Arzneimittel versehentlich eingefroren wurde oder Sie glauben, dass dies passiert sein könnte.
Umstellung von intravenöser auf subkutane Verabreichung
Sobald die Situation unter Kontrolle ist, werden Sie Retacrit weiterhin in regelmäßigen Dosierungen einnehmen. Ihr Arzt kann entscheiden, dass Retacrit als Injektion unter die Haut (subkutan) und nicht in eine Vene (intravenös) verabreicht wird.
Wenn Sie von einer Verabreichungsmethode zur anderen wechseln, muss die Dosierung nicht geändert werden, Ihr Arzt kann dann Blutuntersuchungen anordnen, um zu überprüfen, ob eine Dosisanpassung erforderlich ist.
Spritzen Sie sich Retacrit unter die Haut
Zu Beginn der Behandlung wird Retacrit normalerweise von einem Arzt oder dem medizinischen Fachpersonal verabreicht. Danach kann Ihr Arzt vorschlagen, dass Sie oder Ihr Pflegepersonal lernen, wie man unter die Haut (subkutan) injiziert.
- Versuchen Sie nicht, sich selbst zu injizieren, wenn Ihr Arzt oder das medizinische Fachpersonal Ihnen nicht gesagt hat, wie.
- Wenden Sie Retacrit immer nach Anweisung Ihres Arztes oder des medizinischen Fachpersonals an.
- Verwenden Sie das Arzneimittel nur, wenn es richtig gelagert wurde (siehe Abschnitt 5).
- Nehmen Sie die Spritze vor der Anwendung aus dem Kühlschrank und lassen Sie sie Raumtemperatur annehmen.Dies dauert normalerweise 15-30 Minuten.
Verwenden Sie eine Einzeldosis Retacrit aus jeder Spritze.
Wenn das Arzneimittel unter die Haut (subkutan) verabreicht wird, beträgt das Volumen für eine einzelne Injektion normalerweise nicht mehr als 1 ml.
Retacrit muss allein verabreicht werden und darf nicht mit anderen Injektionsflüssigkeiten gemischt werden.
Schütteln Sie die Fertigspritzen nicht. Längeres und heftiges Schütteln kann das Arzneimittel beschädigen. Verwenden Sie das Arzneimittel nicht, wenn es kräftig geschüttelt wurde.
So injizieren Sie sich mit den Fertigspritzen
- Nehmen Sie die Spritze aus dem Kühlschrank. Die Flüssigkeit muss Raumtemperatur erreichen. Entfernen Sie nicht die Spritzennadelabdeckung, da sie Raumtemperatur erreicht.
- Überprüfen Sie die Spritze, um sicherzustellen, dass sie die richtige Dosis ist, dass sie nicht abgelaufen ist, dass sie nicht beschädigt ist und dass die Flüssigkeit klar und nicht gefroren ist
- Wählen Sie die Injektionsstelle. Die am besten geeigneten Injektionsstellen sind der Oberschenkel und der Bauch, mit Ausnahme des Bereichs um den Nabel. Wechseln Sie jedes Mal die Injektionsstelle.
- Wasch deine Hände. Verwenden Sie ein antiseptisches Tuch, um die Injektionsstelle zu desinfizieren.
- Halten Sie die Spritze mit der abgedeckten Nadel nach oben am Körper.
- Halten Sie die Spritze nicht am Kolbenkopf, Kolben oder Nadelschutz fest.
- Ziehen Sie den Kolben niemals zu sich heran.
- Entfernen Sie die Nadelschutzkappe der Fertigspritze erst, wenn Sie bereit sind, Retacrit zu injizieren.
- Entfernen Sie die Abdeckung von der Spritze, indem Sie den Zylinder festhalten und vorsichtig an der Abdeckung ziehen, ohne sie zu verdrehen. Drücken Sie nicht auf den Kolben, berühren Sie die Nadel nicht und schütteln Sie die Spritze nicht.
- Nehmen Sie eine Hautfalte zwischen Daumen und Zeigefinger, ohne sie zu stark zu drücken.
- Drücken Sie die Nadel ganz hinein.Ihr Arzt oder das medizinische Fachpersonal wird Ihnen gezeigt haben, wie es geht.
- Drücken Sie den Kolben mit dem Daumen ganz hinein, um die gesamte Flüssigkeitsmenge zu injizieren. Drücken Sie ihn langsam und gleichmäßig, wobei Sie die Haut zusammendrücken.
- Wenn der Kolben bis zum Anschlag gedrückt ist, ziehen Sie die Nadel heraus und lassen Sie die Haut los.
- Wenn die Nadel aus der Haut entfernt wird, kann etwas Blut aus der Injektionsstelle austreten.Dies ist normal.Sie können die Injektionsstelle desinfizieren, indem Sie nach der Injektion einige Sekunden lang auf das antiseptische Tuch drücken.
- Legen Sie die gebrauchte Spritze in einen durchstichsicheren Behälter. Versuchen Sie nicht, die Schutzkappe wieder auf die Nadel zu setzen.
- Werfen Sie gebrauchte Spritzen niemals in den Hausmüll.
Verwendung des Nadelschutzes
Die Fertigspritze kann mit einer Sicherheitsvorrichtung für die Nadel ausgestattet werden, die vor versehentlichem Nadelstich schützt.
- Führen Sie die Injektion gemäß der oben beschriebenen Technik durch.
- Halten Sie die Spritze mit den Fingern auf der Auflagekante fest und üben Sie Druck auf den Kolben aus, bis die Injektion der gesamten Dosis abgeschlossen ist. Das Nadelschutzsystem wird NICHT aktiviert, wenn nicht die VOLLE Dosis verabreicht wurde.
- Entfernen Sie die Nadel von Ihrer Haut, lassen Sie dann den Kolben los und die Spritze bewegt sich vorwärts, bis der Schutz die Nadel bedeckt und einrastet.
Wenn Sie die Anwendung von Retacrit vergessen haben
Verwenden Sie nicht die doppelte Dosis, um eine vergessene vorherige Dosis nachzuholen.
Wenn Sie die Einnahme von Retacrit® abbrechen
Brechen Sie die Behandlung nicht ohne vorherige Rücksprache mit Ihrem Arzt ab.
Wenn Sie weitere Fragen zur Anwendung von Retacrit haben, wenden Sie sich an Ihren Arzt.
Überdosierung Was ist zu tun, wenn Sie zu viel Retacrit® eingenommen haben?
Retacrit hat einen großen Sicherheitsspielraum und Nebenwirkungen einer Überdosierung dieses Arzneimittels sind unwahrscheinlich. Informieren Sie sofort Ihren Arzt oder das medizinische Fachpersonal, wenn Sie glauben, dass Sie zu viel Retacrit injiziert haben.
Nebenwirkungen Was sind die Nebenwirkungen von Retacrit®
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Informieren Sie sofort Ihren Arzt, wenn Sie Kopfschmerzen haben, insbesondere wenn es sich um plötzliche, scharfe, migräneähnliche Kopfschmerzen handelt, wenn Sie sich verwirrt fühlen oder Anfälle haben.Diese Symptome können Warnzeichen für einen plötzlichen Anstieg sein.Blutdruck, der einen Notfall erfordert Behandlung.
Informieren Sie Ihren Arzt oder das medizinische Fachpersonal, wenn Sie eine der Wirkungen auf dieser Liste bemerken.
Sehr häufige Nebenwirkungen
Diese können mehr als 1 von 10 mit Retacrit behandelten Personen betreffen.
- Grippeähnliche Symptome, Kopfschmerzen, Gelenkschmerzen, Schwächegefühl, Müdigkeit und Schwindel.
- Bei Patienten mit Nierenerkrankungen, die sich noch keiner Dialyse unterziehen, wurde über eine Verstopfung der Atemwege, wie verstopfte Nase und Halsschmerzen, berichtet.
Häufige Nebenwirkungen
Diese können bis zu 1 von 10 mit Retacrit behandelten Personen betreffen.
- Erhöhter Blutdruck. Der Anstieg des Blutdrucks kann eine Behandlung mit Arzneimitteln erfordern (oder eine Anpassung der Arzneimittel, die Sie bereits wegen Ihres Bluthochdrucks einnehmen). Ihr Arzt wird Ihren Blutdruck während der Behandlung mit Retacrit, insbesondere während der Behandlung mit Retacrit, in regelmäßigen Abständen überprüfen. Beginn der Therapie.
- Brustschmerzen, Kurzatmigkeit, schmerzhafte Schwellungen in den Beinen, die ein Symptom von Blutgerinnseln sein können (Lungenembolie, tiefe Venenthrombose).
- Schlaganfall (unzureichende Blutversorgung des Gehirns, was dazu führen kann, dass eine oder mehrere Gliedmaßen auf einer Körperseite nicht bewegt werden können, nicht verstanden oder gesprochen werden kann oder eine Seite des Gesichtsfelds nicht gesehen wird).
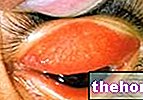
- Hautausschlag und Schwellungen um die Augen (Ödeme), die durch eine allergische Reaktion verursacht werden können.
- Gerinnung in der künstlichen Niere.
Gelegentliche Nebenwirkungen
Diese können bis zu 1 von 100 mit Retacrit behandelten Personen betreffen.
- Hirnblutung.
Seltene Nebenwirkungen
Diese können bis zu 1 von 1.000 mit Retacrit behandelten Personen betreffen.
- Überempfindlichkeitsreaktionen.
Sehr seltene Nebenwirkungen
Diese können bis zu 1 von 10.000 mit Retacrit behandelten Personen betreffen.
- Es kann zu einem Anstieg der Thrombozytenwerte kommen, die normalerweise an der Bildung von Blutgerinnseln beteiligt sind. Der Arzt wird diese Werte überprüfen.
Nebenwirkungen mit nicht bekannter Häufigkeit
Die Häufigkeit dieser Nebenwirkungen kann aus den verfügbaren Daten nicht berechnet werden.
- Schwellungen, insbesondere im Augen- und Lippenbereich (Quincke-Ödem) und schockartige allergische Reaktionen mit Symptomen wie Kribbeln, Rötung, Juckreiz, Rötung und schneller Puls.
- Gefäß- und thrombotische Ereignisse (Blutgerinnsel) in Blutgefäßen wie z. B. eingeschränkte Durchblutung des Gehirns, Netzhautthrombose, eingeschränkte Durchblutung des Herzens, Herzinfarkt, arterielle Thrombose, Erweiterung der Blutgefäßwände (Aneurysma).
- Rote-Serie-Aplasie (PRCA) PRCA wurde bei Patienten nach Monaten bis Jahren einer subkutanen (Injektion unter die Haut) Behandlung mit Erythropoietin berichtet. PRCA bedeutet die Unfähigkeit, eine ausreichende Anzahl roter Blutkörperchen im Knochenmark zu produzieren (siehe Abschnitt „Warnhinweise und Vorsichtsmaßnahmen“).
- Juckreiz.
Andere Nebenwirkungen
Patienten mit Nierenerkrankungen
- Anstieg des Blutdrucks, der eine medikamentöse Behandlung oder eine Dosisanpassung von Arzneimitteln erforderlich machen kann, die Sie bereits gegen Bluthochdruck einnehmen. Ihr Arzt kann Ihren Blutdruck während der Behandlung mit Retacrit regelmäßig messen, insbesondere zu Beginn der Therapie.
- Ein Verschluss der Verbindung zwischen Arterie und Vene (Shuntthrombose) kann insbesondere bei niedrigem Blutdruck oder Komplikationen einer arteriovenösen Fistel auftreten.Ihr Arzt kann den Shunt überprüfen und ein Arzneimittel zur Vorbeugung einer Thrombose verschreiben.
Patienten mit bösartigen Tumoren
- Blutgerinnung (vaskuläre thrombotische Ereignisse) (siehe Abschnitt „Warnhinweise und Vorsichtsmaßnahmen“).
- Erhöhter Blutdruck. Aus diesem Grund sollten Hämoglobinspiegel und Blutdruck überwacht werden.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker oder das medizinische Fachpersonal.Dies gilt auch für alle Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, einschließlich aller möglichen Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem melden. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
Ablauf und Aufbewahrung
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Verwenden Sie dieses Arzneimittel nicht nach dem auf dem Etikett und der Verpackung angegebenen Verfallsdatum ("EXP" / "EXP").
Das Ablaufdatum bezieht sich auf den letzten Tag des Monats.
Im Kühlschrank lagern (2 ° C - 8 ° C). Nicht einfrieren.
Die Spritze kann aus dem Kühlschrank entnommen und einmalig bis zu 3 Tage bei Raumtemperatur belassen werden (jedoch nicht über 25 °C).
Bewahren Sie die Fertigspritze im Umkarton auf, um das Arzneimittel vor Licht zu schützen.
Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker, wie Sie Arzneimittel entsorgen, die Sie nicht mehr verwenden. Dies trägt zum Schutz der Umwelt bei.
Frist "> Weitere Informationen
Was Retacrit enthält
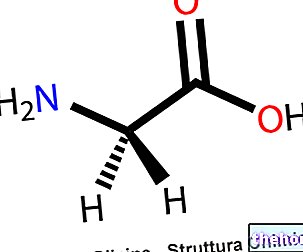
Der Wirkstoff ist Epoetin zeta (hergestellt durch rekombinante DNA-Technik in Ovarialzelllinien des chinesischen Hamsters).
Retacrit 1 000 IE / 0,3 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,3 ml Injektionslösung enthält 1 000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 3 333 IE Epoetin zeta pro ml.
Retacrit 2 000 IE / 0,6 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,6 ml Injektionslösung enthält 2.000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 3 333 IE Epoetin zeta pro ml.
Retacrit 3.000 IE / 0,9 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,9 ml Injektionslösung enthält 3.000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 3 333 IE Epoetin zeta pro ml.
Retacrit 4 000 IE / 0,4 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,4 ml Injektionslösung enthält 4 000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 10 000 IE Epoetin zeta pro ml.
Retacrit 5 000 IE / 0,5 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,5 ml Injektionslösung enthält 5000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 10 000 IE Epoetin zeta pro ml.
Retacrit 6 000 IE / 0,6 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,6 ml Injektionslösung enthält 6 000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 10 000 IE Epoetin zeta pro ml.
Retacrit 8 000 IE / 0,8 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,8 ml Injektionslösung enthält 8 000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 10 000 IE Epoetin zeta pro ml.
Retacrit 10 000 IE / 1 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 1,0 ml Injektionslösung enthält 10 000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 10 000 IE Epoetin zeta pro ml.
Retacrit 20 000 IE / 0,5 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,5 ml Injektionslösung enthält 20 000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 40.000 IE Epoetin zeta pro ml.
Retacrit 30 000 IE / 0,75 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 0,75 ml Injektionslösung enthält 30.000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 40.000 IE Epoetin zeta pro ml.
Retacrit 40 000 IE / 1 ml Injektionslösung in einer Fertigspritze
1 Fertigspritze mit 1,0 ml Injektionslösung enthält 40.000 Internationale Einheiten (IE) Epoetin zeta (rekombinantes humanes Erythropoietin). Die Lösung enthält 40.000 IE Epoetin zeta pro ml. Die sonstigen Bestandteile sind Dinatriumphosphat-Dihydrat, einbasisches Natriumphosphat-Dihydrat, Natriumchlorid, Calciumchlorid-Dihydrat, Polysorbat 20, Glycin, Leucin, Isoleucin, Threonin, Glutaminsäure, Phenylalanin und Wasser für Injektionszwecke, Natriumhydroxid (zur pH-Einstellung), Salzsäure (um den pH-Wert einzustellen).
Wie Retacrit aussieht und Inhalt der Packung
Retacrit ist eine klare und farblose Injektionslösung in klaren, farblosen Glasspritzen mit fester Nadel.
Die Fertigspritzen enthalten je nach Epoetin zeta-Gehalt 0,3 ml bis 1 ml Lösung (siehe Abschnitt „Was Retacrit enthält“).
Eine Packung enthält 1 oder 4 oder 6 Fertigspritzen mit oder ohne Nadelschutz.
Bündelpackungen enthalten 4 (4 Packungen mit 1) oder 6 (6 Packungen mit 1) Fertigspritzen.
Quelle Packungsbeilage: AIFA (Italienische Arzneimittelbehörde). Im Januar 2016 veröffentlichter Inhalt. Die vorliegenden Informationen können nicht aktuell sein.
Um Zugriff auf die aktuellste Version zu haben, ist es ratsam, auf die Website der AIFA (Italienische Arzneimittelbehörde) zuzugreifen. Haftungsausschluss und nützliche Informationen.
01.0 BEZEICHNUNG DES ARZNEIMITTELS -
RETACRIT 1000 IE / 0,3 ML LÖSUNG ZUR INJEKTION IN FERTIGSPRITZE
02.0 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG -
Eine Fertigspritze mit 0,3 ml Injektionslösung enthält 1 000 Internationale Einheiten (IE) Epoetin zeta* (rekombinantes humanes Erythropoietin). Die Lösung enthält 3 333 IE Epoetin zeta pro ml.
* Produziert durch rekombinante DNA-Technik in Zellinien der Eierstöcke des Chinesischen Hamsters (CHO).
Sonstiger Bestandteil mit bekannter Wirkung:
Jede Fertigspritze enthält 0,15 mg Phenylalanin.
Die vollständige Auflistung der sonstigen Bestandteile finden Sie in Abschnitt 6.1.
03.0 DARREICHUNGSFORM -
Injektionslösung in einer Fertigspritze.
Klare und farblose Lösung.
04.0 KLINISCHE INFORMATIONEN -
04.1 Anwendungsgebiete -
• Behandlung von symptomatischer Anämie im Zusammenhang mit chronischem Nierenversagen (CRI) bei erwachsenen und pädiatrischen Patienten:
• Behandlung von Anämie im Zusammenhang mit chronischem Nierenversagen bei erwachsenen und pädiatrischen Patienten unter Hämodialyse und bei erwachsenen Patienten unter Peritonealdialyse (siehe Abschnitt 4.4).
• Behandlung einer schweren renalen Anämie mit klinischen Symptomen bei erwachsenen Patienten mit Niereninsuffizienz, die sich noch nicht einer Dialyse unterziehen (siehe Abschnitt 4.4).
- Behandlung von Anämie und Reduzierung des Transfusionsbedarfs bei erwachsenen Patienten, die sich einer Chemotherapie wegen solider Tumore, eines malignen Lymphoms oder eines multiplen Myeloms unterziehen und bei denen das Risiko einer Bluttransfusion entsprechend dem Allgemeinzustand des Patienten besteht (Herz-Kreislauf-Situation, bereits bestehende Anämie zu Beginn der Chemotherapie) .
- Retacrit kann zur Erhöhung der Eigenblutmenge bei Patienten verwendet werden, die an einem Blutspendeprogramm teilnehmen. Die Anwendung in dieser Indikation sollte unter Berücksichtigung der berichteten Risiken thromboembolischer Ereignisse beurteilt werden Die Behandlung sollte nur Patienten mit mittelschwerer Anämie (ohne Eisenmangel) vorbehalten sein, wenn blutkonservierende Verfahren nicht verfügbar oder unzureichend sind, wenn „geplante größere elektive chirurgische Eingriffe erforderlich“ sind eine große Blutmenge (4 oder mehr Einheiten Blut für Frauen, 5 oder mehr Einheiten für Männer).
- Retacrit kann verwendet werden, um die Exposition gegenüber allogenen Bluttransfusionen bei erwachsenen Patienten ohne Eisenmangel zu reduzieren, von denen angenommen wird, dass sie ein hohes Risiko für Transfusionskomplikationen vor größeren elektiven orthopädischen Operationen haben ), die nicht Teil eines Eigenblutspendeprogramms sind und für die ein mäßiger Blutverlust erwartet wird (von 900 bis 1 800 ml).
04.2 Dosierung und Art der Anwendung -
Die Therapie mit Retacrit sollte unter Aufsicht von medizinischem Personal eingeleitet werden, das Erfahrung in der Behandlung von Patienten mit den oben beschriebenen Indikationen hat.
Dosierung
Behandlung symptomatischer Anämie bei erwachsenen und pädiatrischen Patienten mit chronischem Nierenversagen
Retacrit sollte entweder subkutan oder intravenös verabreicht werden.
Die gewünschte Hämoglobinkonzentration liegt zwischen 10 und 12 g/dl (6,2-7,5 mmol/l), außer bei pädiatrischen Patienten, wo die Hämoglobinkonzentration zwischen 9,5 und 11 g/dl (5,9-6,8 mmol/l) liegen muss. Die Obergrenze der Hämoglobin-Zielkonzentration sollte nicht überschritten werden. Symptome und Folgen einer Anämie können je nach Alter, Geschlecht und allgemeiner Krankheitslast variieren; Es ist notwendig, dass der klinische Verlauf und der Zustand des einzelnen Patienten vom Arzt beurteilt werden. Retacrit sollte entweder subkutan oder intravenös verabreicht werden, um Hämoglobinwerte von nicht mehr als 12 g / dl (7,5 mmol / l) zu erreichen. Aufgrund der Variabilität zwischen den Patienten können bei einem Patienten gelegentlich einzelne Hämoglobinwerte oberhalb und unterhalb der gewünschten Hämoglobinkonzentration beobachtet werden. Schwankungen des Hämoglobins sollten durch Dosisanpassung gesteuert werden, bezogen auf einen Hämoglobin-Zielbereich zwischen 10 g / dl (6,2 mmol / l) und 12 g / dl (7,5 mmol / l).
Ein längerer Hämoglobinspiegel über 12 g/dl (7,5 mmol/l) sollte vermieden werden; Anweisungen für eine angemessene Dosisanpassung, wenn Hämoglobinwerte über 12 g / dl (7,5 mmol / l) beobachtet werden, sind unten angegeben. Ein Anstieg des Hämoglobins von mehr als 2 g/dl (1,25 mmol/l) über einen Zeitraum von vier Wochen sollte vermieden werden. In diesem Fall sollte eine geeignete Dosisanpassung wie angegeben vorgenommen werden.
Die Patienten sollten engmaschig überwacht werden, um sicherzustellen, dass die niedrigste zugelassene wirksame Dosis von Retacrit angewendet wird, um die Symptome einer Anämie durch Aufrechterhaltung einer Hämoglobinkonzentration von weniger als oder gleich 12 g / dl (7,5 mmol / l) angemessen zu kontrollieren.
Bei Patienten mit chronischer Niereninsuffizienz ist bei der Erhöhung der Retacrit-Dosen Vorsicht geboten.Bei Patienten mit schwacher Hämoglobin-Reaktion auf Retacrit sollten alternative Erklärungen für diese schwache Reaktion in Betracht gezogen werden (siehe Abschnitte 4.4 und 5.1).Bei Patienten mit chronischer Niereninsuffizienz Krankheit und klinischen Anzeichen einer ischämischen Herzkrankheit oder einer kongestiven Herzinsuffizienz sollte die Hämoglobinkonzentration zur Erhaltungstherapie die maximale Zielkonzentrationsgrenze nicht überschreiten.
Erwachsene Hämodialysepatienten
Retacrit sollte entweder subkutan oder intravenös verabreicht werden.
Die Behandlung gliedert sich in zwei Phasen:
1. Korrekturphase: 50 IE / kg, 3-mal pro Woche. Wenn eine Dosisanpassung erforderlich ist, sollte dies schrittweise in Abständen von mindestens 4 Wochen erfolgen. Bei jeder Anpassung sollte die Dosis dreimal pro Woche um 25 IE / kg erhöht oder verringert werden.
2. Erhaltungsphase: Dosisanpassung zur Aufrechterhaltung des gewünschten Hämoglobinspiegels (Hb) zwischen 10 und 12 g / dl (6,2 - 7,5 mmol / l). Die empfohlene wöchentliche Gesamtdosis reicht von 75 bis 300 IE / kg.
Verfügbare klinische Daten weisen darauf hin, dass Patienten mit einem sehr niedrigen anfänglichen Hämoglobinspiegel (8 g / dl oder > 5 mmol / l).
Pädiatrische Patienten unter Hämodialyse
Die Behandlung gliedert sich in zwei Phasen.
1. Korrekturphase 50 IE/kg, 3 mal pro Woche intravenös. Falls eine Dosisanpassung erforderlich ist, sollte diese in Schritten von 25 I.E./kg 3-mal pro Woche in Abständen von mindestens 4 Wochen erfolgen, bis das Ziel erreicht ist.
2. Erhaltungsphase Dosisanpassung zur Aufrechterhaltung des gewünschten Hämoglobinspiegels (Hb) zwischen 9,5 und 11 g / dl (5,9-6,8 mmol / l).
Im Allgemeinen benötigen Kinder und Jugendliche mit einem Körpergewicht unter 30 kg höhere Erhaltungsdosen als Kinder mit einem Körpergewicht über 30 kg und Erwachsene. In klinischen Studien wurden beispielsweise nach 6-monatiger Behandlung die folgenden Erhaltungsdosen beobachtet:
Verfügbare klinische Daten weisen darauf hin, dass Patienten mit einem sehr niedrigen anfänglichen Hämoglobinspiegel (6,8 g / dl oder > 4,25 mmol / l) sind.
Erwachsene Patienten mit Peritonealdialyse
Retacrit sollte entweder subkutan oder intravenös verabreicht werden.
Die Behandlung gliedert sich in zwei Phasen.
1. Korrekturphase: Die Anfangsdosis beträgt 50 IE / kg Körpergewicht zweimal wöchentlich.
2. Erhaltungsphase: Dosisanpassung zur Aufrechterhaltung des gewünschten Hämoglobinspiegels (Hb), (zwischen 10 und 12 g / dl [6,2-7,5 mmol / l]. Die Erhaltungsdosis beträgt zwischen 25 und 50 IE / kg 2-mal a Woche, aufgeteilt in 2 gleiche Dosen.
Erwachsene Patienten mit Niereninsuffizienz, die noch keine Dialyse erhalten
Retacrit sollte entweder subkutan oder intravenös verabreicht werden.
Die Behandlung gliedert sich in zwei Phasen.
1. Korrekturphase: Eine Anfangsdosis von 50 I.E./kg 3-mal pro Woche, ggf. gefolgt von einer Erhöhung in Schritten von 25 I.E./kg (3-mal pro Woche) bis das gewünschte Ziel erreicht ist (die Erhöhung sollte schrittweise erfolgen, im Abstand von mindestens vier Wochen).
2. Erhaltungsphase: Während der Erhaltungsphase kann Retacrit 3-mal wöchentlich und bei subkutaner Gabe einmal wöchentlich oder alle zwei Wochen verabreicht werden. Die Dosis und die Dosierungsintervalle müssen korrekt angepasst werden, um den gewünschten Hämoglobin (Hb)-Spiegel aufrechtzuerhalten (zwischen 10 und 12 g / dl [6,2-7,5 mmol / l].
Eine Verlängerung des Dosierungsintervalls kann eine Dosiserhöhung erfordern.
Die Höchstdosis sollte 3 mal wöchentlich 150 IE/kg, einmal wöchentlich 240 IE/kg (bis maximal 20.000 IE) oder einmal wöchentlich 480 IE/kg (bis maximal 40.000 IE) nicht überschreiten . alle 2 Wochen.
Behandlung von Patienten mit Chemotherapie-induzierter Anämie
Retacrit sollte bei anämischen Patienten (zB mit Hämoglobinkonzentration ≤ 10 g/dl (6,2 mmol/l) subkutan verabreicht werden. Die Symptome und Folgen einer Anämie können je nach Alter, Geschlecht und Schweregrad variieren. Gesamterkrankung; eine individuelle Beurteilung der klinischen Verlauf und Zustand jedes einzelnen Patienten wird vom Arzt gefordert.
Angesichts der Intra-Patienten-Variabilität können bei einem Patienten gelegentlich einzelne Hämoglobinwerte oberhalb und unterhalb des gewünschten Hämoglobinspiegels nachgewiesen werden. Die Hämoglobinvariabilität sollte durch Dosisanpassung relativ zu einem Hämoglobin-Zielbereich von 10 g/dl (6,2 mmol/l) bis 12 g/dl (7,5 mmol/l) gesteuert werden. Ein längerer Hämoglobinspiegel über 12 g/dl (7,5 mmol/l) sollte vermieden werden; Anweisungen für eine angemessene Dosisanpassung, wenn Hämoglobinwerte über 12 g / dl (7,5 mmol / l) beobachtet werden, sind unten angegeben.
Die Patienten sollten engmaschig überwacht werden, um sicherzustellen, dass die niedrigste zugelassene Dosis von Retacrit angewendet wird, um die Symptome einer Anämie angemessen zu kontrollieren.
Die Retacrit-Therapie sollte nach Beendigung der Chemotherapie noch einen Monat lang fortgesetzt werden. Die Anfangsdosis beträgt 150 IE / kg, 3-mal pro Woche subkutan. Alternativ kann Retacrit in der Anfangsdosis von 450 IE/kg einmal wöchentlich subkutan verabreicht werden. Wenn nach 4-wöchiger Behandlung das Hämoglobin um mindestens 1 g/dl (0,62 mmol/l) angestiegen ist oder die Retikulozytenzahl um ≥ 40.000 Zellen/μl gegenüber dem Ausgangswert angestiegen ist, sollte die Dosis einmal wöchentlich bei 450 IE/kg bleiben oder 150 IE / kg 3 mal pro Woche Wenn der Hämoglobinanstieg
Das empfohlene Dosierungsschema ist in der folgenden Tabelle aufgeführt:
Sobald das therapeutische Ziel für den einzelnen Patienten erreicht ist, sollte die Dosis um 25 bis 50 % reduziert werden, um das Hämoglobin auf diesem Niveau zu halten. Eine geeignete Dosistitration sollte in Betracht gezogen werden.
Dosisanpassung
Wenn der Anstieg des Hämoglobins mehr als 2 g / dl (> 1,25 mmol / l) pro Monat beträgt, sollte die Dosis von Retacrit um ca. 25-50% reduziert werden.Wenn der Hämoglobinwert 12 g / dl (7,5 mmol / l) überschreitet ), unterbrechen Sie die Therapie, bis sie auf 12 g / dl (7,5 mmol / l) zurückgekehrt ist oder unter diese abfällt, und setzen Sie dann die Retacrit-Therapie mit einer Dosis von weniger als 25 % der vorherigen Dosis fort.
Behandlung von erwachsenen Patienten, die Kandidaten für chirurgische Eingriffe im Rahmen von Eigenblutspendeprogrammen sind
Retacrit muss intravenös verabreicht werden.
Zum Zeitpunkt der Blutspende muss Retacrit nach Abschluss des Spendeverfahrens verabreicht werden.
Patienten mit leichter Anämie (Hämatokrit 33-39%), die eine Vorablagerung von ≥ 4 Einheiten Blut benötigen, sollten 3 Wochen vor der Operation 2 mal pro Woche mit 600 IE/kg Retacrit behandelt werden.
Während der gesamten Dauer der Retacrit-Therapie sollten alle Patienten eine ausreichende Eisenergänzung erhalten (z. B. 200 mg / Tag orales elementares Eisen). Mit der Eisengabe sollte so früh wie möglich begonnen werden, auch mehrere Wochen vor der Durchführung der autologen Prädeposition, um die Eisenspeicher vor Beginn der Therapie mit Retacrit aufzustocken.
Behandlung von erwachsenen Patienten, die für einen größeren elektiven orthopädischen Eingriff vorgesehen sind
Retacrit sollte subkutan verabreicht werden.
Drei Wochen (Tage 21, 14 und 7) vor der Operation und am Operationstag (Tag 0) sollte einmal wöchentlich eine Dosis von 600 IE/kg Körpergewicht verabreicht werden. In Fällen, in denen die Zeit vor der Operation auf weniger als drei Wochen verkürzt werden muss, sollte an 10 aufeinander folgenden Tagen vor der Operation, am Tag der Operation und in den vier Tagen eine Tagesdosis von 300 IE / kg Körpergewicht verabreicht werden der Hämoglobinspiegel im Rahmen hämatologischer Tests in der präoperativen Phase 15 g / dl erreicht oder überschreitet, sollte Retacrit abgesetzt und nachfolgende Dosen nicht verabreicht werden.
Eisenmangel muss vor Beginn der Behandlung mit Retacrit behandelt werden. Darüber hinaus sollte allen mit Retacrit behandelten Patienten während der gesamten Dauer der Behandlung mit Retacrit eine ausreichende Eisenmenge (z. B. 200 mg Eisenionen oral pro Tag) vor der Retacrit-Therapie verabreicht werden, um ausreichende Ablagerungen zu erzielen.
Art der Verabreichung
Intravenöse Injektion
Die Verabreichung sollte je nach Gesamtdosis mindestens 1-5 Minuten dauern. Bei Hämodialysepatienten ist es möglich, die Bolusdosis während der Dialysesitzung über einen geeigneten venösen Zugang des Dialysekreislaufs zu verabreichen. Alternativ kann die Substanz am Ende der Dialysesitzung durch die Fistel injiziert werden und gefolgt von 10 ml 9 mg/ml (0,9 %) physiologischer NaCl-Lösung, um den Kreislauf zu spülen und eine zufriedenstellende Einführung des Produkts in den Kreislauf zu gewährleisten. Bei Patienten, die auf die Behandlung mit grippeähnlichen Symptomen reagieren, ist eine langsamere Anwendung vorzuziehen.
Retacrit darf nicht als intravenöse Infusion verabreicht werden.
Retacrit darf nicht mit anderen Arzneimitteln gemischt werden (siehe Abschnitt 6.2).
Subkutane Injektion
Generell sollte das maximale Volumen von 1 ml pro Injektionsstelle nicht überschritten werden, bei größeren Volumina müssen mehrere Applikationsstellen gewählt werden.
Injektionen werden in die Gliedmaßen oder in die vordere Bauchdecke verabreicht.
Hinweise zur Handhabung des Arzneimittels vor der Anwendung siehe Abschnitt 6.6.
04.3 Kontraindikationen -
- Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
- Patienten mit einer Erythropoietin-Behandlung mit reiner Erythrocyten-Aplasie (PRCA) sollten sich keiner Therapie mit Retacrit oder einem anderen Erythropoietin-Typ unterziehen (siehe Abschnitt 4.4).
- Unkontrollierter Bluthochdruck.
- Bei der Indikation „Erhöhung der Eigenblutmenge“: Myokardinfarkt oder Schlaganfall im Monat vor der Behandlung, instabile Angina pectoris, erhöhtes Risiko für eine tiefe Venenthrombose, wie eine thromboembolische Venenerkrankung in der Vorgeschichte.
- Bei der Indikation für einen größeren elektiven orthopädischen Eingriff: schwere koronare, periphere arterielle, karotis- oder zerebrale Gefäßerkrankungen, einschließlich Patienten mit kürzlich aufgetretenem Myokardinfarkt oder zerebrovaskulärem Unfall.
- Patienten, die aus irgendeinem Grund keine "adäquate antithrombotische Prophylaxe" erhalten können.
04.4 Besondere Warnhinweise und geeignete Vorsichtsmaßnahmen für die Anwendung -
Allgemeine Information
Wie bei allen Patienten, die Erythropoietin erhalten, kann es während der Behandlung mit Retacrit zu einem Blutdruckanstieg kommen. Sowohl bei allen Patienten, die zum ersten Mal mit Epoetin behandelt werden, als auch bei Patienten, die bereits behandelt wurden, sollte der Blutdruck vor, zu Beginn und während der Behandlung mit Retacrit sorgfältig überwacht und angemessen kontrolliert werden -hypertensive Behandlung Wenn der Blutdruck nicht kontrolliert werden kann, sollte die Behandlung mit Retacrit abgebrochen werden.
Retacrit sollte auch bei Epilepsie und chronischem Leberversagen mit Vorsicht angewendet werden.
Während der Behandlung mit Erythropoietin kann ein mäßiger dosisabhängiger Anstieg der Thrombozytenzahl innerhalb des Normbereichs auftreten. Dieses Phänomen bildet sich mit der Fortsetzung der Therapie zurück. Es wird empfohlen, die Thrombozytenzahl während der ersten 8 Wochen der Therapie regelmäßig zu kontrollieren.
Alle anderen Ursachen einer Anämie (Eisenmangel, Hämolyse, Blutverlust, Vitamin B12- oder Folsäuremangel) sollten vor und während der Behandlung mit Retacrit untersucht und behandelt werden. In den meisten Fällen sinken die Serum-Ferritin-Werte gleichzeitig mit den Hämatokrit-Werten.Um ein optimales Ansprechen auf Erythropoietin zu gewährleisten, müssen ausreichende Eisenspeicher sichergestellt werden:
- bei Patienten mit chronischer Niereninsuffizienz und Serum-Ferritinspiegeln unter 100 ng/ml wird eine Eisensupplementierung empfohlen, zum Beispiel 200-300 mg/Tag oral (100-200 mg/Tag bei pädiatrischen Patienten);
- Bei allen Krebspatienten mit Transferrin-Sättigungswerten unter 20% wird eine orale Eisenergänzung von 200-300 mg/Tag empfohlen.
Alle diese Faktoren, die zum Auftreten einer Anämie beitragen, müssen ebenfalls sorgfältig abgewogen werden, bevor eine Erhöhung der Erythropoietin-Dosis bei Krebspatienten beschlossen wird.
Ein paradoxer Abfall des Hämoglobins und die Entwicklung einer schweren Anämie in Verbindung mit niedrigen Retikulozytenzahlen sollten sie darauf aufmerksam machen, die Behandlung mit Epoetin abzubrechen und Anti-Erythropoietin-Antikörpertests durchzuführen.Bei Patienten mit Hepatitis C, die mit Interferon und Ribavirin behandelt wurden, wurden Fälle berichtet, wenn Epoetine wurden in Kombination angewendet.Epoetine sind nicht für die Behandlung von Anämie im Zusammenhang mit Hepatitis C zugelassen.
Um die Rückverfolgbarkeit von Erythropoese-stimulierenden Wirkstoffen (Erythropie-stimulierende Mittel (ESA) muss der Name des verschriebenen ESA in der Krankenakte des Patienten deutlich vermerkt (oder angegeben) werden.
In der perioperativen Behandlung sollte immer eine gute Blutmanagementpraxis angewendet werden.
Patienten, die für einen größeren elektiven orthopädischen Eingriff vorgesehen sind
Bei Patienten, bei denen ein größerer elektiver orthopädischer Eingriff geplant ist, sollten die Ursachen der Anämie ermittelt und behandelt werden, möglicherweise vor Beginn der Behandlung mit Retacrit. Thrombotische Ereignisse können bei dieser Patientenpopulation ein Risiko darstellen, und dies sollte im Hinblick auf den erwarteten Behandlungsnutzen sorgfältig abgewogen werden.Die Patienten sollten eine angemessene antithrombotische Prophylaxe erhalten, da bei chirurgischen Patienten, insbesondere bei Patienten mit kardiovaskulären Grunderkrankungen, thrombotische und vaskuläre Ereignisse auftreten können. Darüber hinaus ist besondere Vorsicht geboten bei Patienten, die für die Entwicklung einer tiefen Venenthrombose (TVT) prädisponiert sind Bei Patienten mit Ausgangshämoglobin > 13 g / dl kann die Möglichkeit nicht ausgeschlossen werden, dass die Behandlung mit Retacrit mit einem erhöhten Risiko für postoperative thrombotische / vaskuläre Ereignisse verbunden ist. Daher sollte eine solche Behandlung bei Patienten mit Ausgangshämoglobin > 13 g / dl nicht angewendet werden .
Patienten mit chronischer Niereninsuffizienz
Hämoglobinkonzentration
Bei Patienten mit chronischer Niereninsuffizienz sollte die Hämoglobin-Erhaltungskonzentration die in Abschnitt 4.2 empfohlene Obergrenze der Hämoglobin-Zielkonzentration nicht überschreiten. In klinischen Studien wurden ein erhöhtes Sterberisiko, schwere kardiovaskuläre Ereignisse und zerebrovaskuläre Ereignisse einschließlich Schlaganfall beobachtet, wenn ESA verabreicht wurde, um Hämoglobinwerte von mehr als 12 g / dl (7,5 mmol / l) zu erreichen.
Kontrollierte klinische Studien haben keinen signifikanten Nutzen gezeigt, der der Verabreichung von Epoetinen zuzuschreiben ist, wenn die Hämoglobinkonzentration die Werte überschritten hat, die zur Kontrolle der Anämiesymptome und zur Vermeidung von Bluttransfusionen erforderlich sind.
Der Hämoglobinspiegel sollte in regelmäßigen Abständen gemessen werden, bis er einen konstanten Wert erreicht, und danach in regelmäßigen Abständen. Der Anstieg des Hämoglobins sollte ungefähr 1 g / dl (0,62 mmol / l) pro Monat betragen und sollte 2 g / dl (1,25 mmol / l) pro Monat nicht überschreiten, um das Risiko einer Hypertonie oder ihrer Verschlimmerung zu minimieren.
Patienten mit chronischer Niereninsuffizienz, die subkutan mit Retacrit behandelt werden, sollten regelmäßig auf einen Wirksamkeitsverlust überwacht werden, definiert als Nichtansprechen oder vermindertes Ansprechen auf die Behandlung mit Retacrit bei Patienten, die zuvor auf eine solche Therapie angesprochen haben. Dies ist durch einen anhaltenden Abfall des Hämoglobins trotz einer Erhöhung der Retacrit-Dosis gekennzeichnet.
Einige Patienten, die mit Epoetin alfa in längeren Dosierungsintervallen (mehr als einmal wöchentlich) behandelt werden, können möglicherweise keine ausreichenden Hämoglobinspiegel aufrechterhalten (siehe Abschnitt 5.1) und erfordern möglicherweise eine Dosiserhöhung. Der Hämoglobinspiegel sollte regelmäßig überwacht werden.
Bei Patienten mit chronischer Niereninsuffizienz ist bei steigenden Retacrit-Dosen Vorsicht geboten, da hohe kumulative Epoetin-Dosen mit einem erhöhten Mortalitätsrisiko und schwerwiegenden kardiovaskulären und zerebrovaskulären Ereignissen verbunden sein können ein schwaches Ansprechen sollte in Betracht gezogen werden (siehe Abschnitte 4.4 und 5.1).
Bei fehlendem Ansprechen auf eine Erythropoietin-Therapie müssen die verantwortlichen Faktoren umgehend untersucht werden. Dazu gehören: Eisen-, Folat- oder Vitamin B12-Mangel, Aluminiumintoxikation, interkurrente Infektionen, entzündliche oder traumatische Episoden, okkulter Blutverlust, Hämolyse, Knochenmarkfibrose jeglicher Herkunft.
Bei Patienten mit chronischem Nierenversagen, die subkutanes Erythropoietin erhielten, wurde sehr selten über Fälle von Antikörper-vermittelter PRCA berichtet. Bei Patienten, die einen „plötzlichen Wirkungsverlust, nachgewiesen durch eine Abnahme des Hämoglobins (1-2 g/dl pro Monat) mit erhöhtem Transfusionsbedarf zeigen, sollte eine Retikulozytenzählung durchgeführt werden und typische Ursachen, die ein Ansprechen auf die Behandlung verhindern (z Folat- oder Vitamin-B12-Mangel, Aluminiumvergiftung, Infektion oder Entzündung, Blutverlust, Hämolyse) Wenn keine Ursache gefunden wird, sollte eine Blutuntersuchung im Knochenmark in Erwägung gezogen werden, um eine PRCA zu diagnostizieren.
Wenn eine PRCA diagnostiziert wird, sollte die Retacrit-Therapie sofort abgebrochen und ein Test auf das Vorhandensein von Anti-Erythropoietin-Antikörpern erwogen werden. Die Patienten sollten nicht auf eine Behandlung mit einem anderen Arzneimittel umgeleitet werden, da eine Kreuzreaktivität zwischen Anti-Erythropoietin-Antikörpern und anderen Erythropoietinen vorliegt Andere Ursachen der PRCA müssen ausgeschlossen und eine geeignete Therapie eingeleitet werden.
Eine regelmäßige Überwachung der Retikulozytenzahl wird empfohlen, um einen Verlust der therapeutischen Wirksamkeit bei Patienten mit chronischem Nierenversagen zu erkennen.
In Einzelfällen wurde eine Hyperkaliämie beobachtet. Bei Patienten mit chronischer Niereninsuffizienz kann die Korrektur der Anämie zu einer Steigerung des Appetits und der Aufnahme von Kalium und Protein führen. Verschriebene Parameter für die Dialyse müssen möglicherweise regelmäßig angepasst werden, um Harnstoff, Kreatinin und Kalium innerhalb der gewünschten Werte zu halten. Bei Patienten mit chronischer Niereninsuffizienz Serum Elektrolyte sollten überwacht werden.Wenn erhöhte (oder steigende) Serumkaliumwerte beobachtet werden, sollte erwogen werden, die Verabreichung von Erythropoietin abzusetzen, bis die Hyperkaliämie korrigiert ist.
Während einer Erythropoietin-Therapie ist aufgrund einer Erhöhung des Hämatokritwertes häufig eine Erhöhung der Heparin-Dosis erforderlich. Wenn die Heparinisierung nicht optimal ist, kann es zu einem Verschluss des Dialysesystems kommen.
Nach den bisher verfügbaren Daten beschleunigt die Korrektur einer Anämie mit Erythropoietin bei erwachsenen Patienten mit Niereninsuffizienz, die sich noch nicht einer Dialyse unterziehen, das Fortschreiten der Niereninsuffizienz nicht.
Erwachsene Krebspatienten mit symptomatischer Anämie unter Chemotherapie
Bei Krebspatienten, die eine Chemotherapie erhalten, sollte das 2-3-wöchige Intervall zwischen der Anwendung und dem Auftreten von Erythropoietin-induzierten Erythrozyten bei der Beurteilung der Angemessenheit einer Retacrit-Therapie berücksichtigt werden (Patienten mit Transfusionsrisiko). mehr als 2 g / dl (1,25 mmol / l) pro Monat oder wenn sein Gehalt 12 g / dl (7,5 mmol / l) überschreitet, ist die Dosisanpassung gemäß Abschnitt 4.2 sorgfältig durchzuführen, um das Potenzial zu reduzieren Risikofaktoren für thrombotische Ereignisse (siehe Abschnitt 4.2).
Da bei Krebspatienten, die mit Erythropoetika behandelt werden, eine erhöhte Inzidenz thromboembolischer Ereignisse beobachtet wurde (siehe Abschnitt 4.8), sollte dieses Risiko im Hinblick auf den Nutzen einer Behandlung (mit Retacrit) sorgfältig abgewogen werden, insbesondere bei Krebspatienten, die ein erhöhtes thromboembolisches Risiko, wie bei adipösen Patienten oder mit einer Vorgeschichte von thrombotischen und vaskulären Ereignissen (tiefe Venenthrombose, Lungenembolie).
Erwachsene Patienten, die für eine Operation in Frage kommen, die Teil eines autologen Blutspendeprogramms ist
Alle Warnhinweise und besonderen Vorsichtsmaßnahmen im Zusammenhang mit autologen Blutspendeprogrammen müssen beachtet werden, insbesondere durch Wiederherstellung der normalen Blutentnahmemenge.
Onkogenes Potenzial
Epoetine sind Wachstumsfaktoren, die in erster Linie die Produktion von Erythrozyten anregen. Erythropoietin-Rezeptoren können auf der Oberfläche einer Reihe von neoplastischen Zellen exprimiert werden. Wie bei allen Wachstumsfaktoren besteht Zweifel, dass Epoetin das Wachstum aller malignen Tumoren stimulieren kann. In mehreren kontrollierten klinischen Studien war dies nicht der Fall. Epoetine haben Es wurde gezeigt, dass es das Gesamtüberleben verbessert oder das Risiko einer Krebsprogression bei Patienten mit krebsassoziierter Anämie verringert.
Mehrere kontrollierte klinische Studien, in denen Epoetine an Patienten mit einer Reihe von häufigen malignen Erkrankungen wie Plattenepithelkarzinomen des Kopf-Hals-Bereichs, Lungenkrebs und Brustkrebs verabreicht wurden, zeigten einen unerklärlichen Anstieg der Sterblichkeitsrate. In kontrollierten klinischen Studien zeigte die Anwendung von Epoetin alfa und anderen Erythropoese-stimulierenden Mitteln (ESAs):
• Eine Verkürzung der Zeit bis zur Tumorprogression bei Patienten mit fortgeschrittenem Kopf-Hals-Karzinom, die mit Strahlentherapie behandelt werden, wenn sie verabreicht werden, um Hämoglobinwerte von mehr als 14 g / dl (8,7 mmol / l) zu erreichen,
• eine Verringerung des Gesamtüberlebens und eine Zunahme der Todesfälle aufgrund der Tumorprogression nach 4 Monaten bei Patientinnen mit metastasiertem Brustkrebs, die mit Chemotherapie behandelt wurden, wenn sie verabreicht wurden, um Hämoglobinwerte von 12-14 g / dl (7,5-8,7 mmol / l) zu erreichen. ,
• ein erhöhtes Sterberisiko bei der Verabreichung von Hämoglobinwerten von 12 g / dl (7,5 mmol / l) bei Patienten mit aktiven Malignomen, die nicht mit Chemotherapie oder Strahlentherapie behandelt werden. Die Anwendung von ESA ist bei dieser Patientenpopulation nicht angezeigt.
Ausgehend davon sollte bei einigen klinischen Zuständen eine Bluttransfusion die bevorzugte Behandlung zur Behandlung einer Anämie bei Krebspatienten sein und sollte den spezifischen klinischen Kontext berücksichtigen. Bei dieser Bewertung sollten die Art des Krebses und sein Stadium, der Grad der Anämie, die Lebenserwartung, die Umgebung, in der der Patient behandelt wird, und die Präferenzen des Patienten berücksichtigt werden (siehe Abschnitt 5.1).
Dieses Arzneimittel enthält Phenylalanin, eine Substanz, die für Menschen mit Phenylketonurie gefährlich sein kann.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, was bedeutet, dass es als „natriumarm“ gilt.
04.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen -
Es wurde nicht gezeigt, dass die Behandlung mit Erythropoietin den Metabolismus anderer Arzneimittel verändert. Da Ciclosporin jedoch an Erythrozyten bindet, kann die Möglichkeit einer "Wechselwirkungen mit anderen Arzneimitteln" bestehen. Bei gleichzeitiger Anwendung von Erythropoietin mit Ciclosporin sollten die Blutspiegel von Ciclosporin überwacht und die Dosis dieses Arzneimittels muss gemäß die Erhöhung des Hämatokritwertes.
Es gibt keine Hinweise auf eine „Wechselwirkung zwischen Epoetin alfa und G-CSF oder GM-CSF im Hinblick auf die hämatologische Differenzierung oder Proliferation in Tumorbiopsien. in vitro.
04.6 Schwangerschaft und Stillzeit -
Es liegen keine adäquaten und gut kontrollierten Studien bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3). Es ist nicht bekannt, ob exogenes Epoetin zeta in die Muttermilch übergeht, daher sollte Erythropoietin während der Schwangerschaft und Stillzeit im Allgemeinen nur angewendet werden, wenn der potenzielle Nutzen die potenziellen Risiken für den Fötus überwiegt.
04.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen -
Retacrit hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
04.8 Nebenwirkungen -
Zusammenfassung des Sicherheitsprofils
Die Ergebnisse klinischer Studien mit Retacrit stimmen mit dem Sicherheitsprofil anderer zugelassener Erythropoietine überein. Basierend auf den Ergebnissen klinischer Studien mit anderen zugelassenen Erythropoietinen wird erwartet, dass bei etwa 8 % der mit Erythropoietin behandelten Patienten Nebenwirkungen auftreten durch Kopfschmerzen und einen dosisabhängigen Blutdruckanstieg Es können hypertensive Krisen mit Symptomen ähnlich einer "Enzephalopathie" auftreten. Auf plötzlich auftretende akute migräneartige Kopfschmerzen sollte geachtet werden, die ein Warnsignal sein können.
In einigen Studien bei erwachsenen Patienten mit Niereninsuffizienz, die sich noch nicht einer Dialyse unterziehen und mit verlängerten Dosierungsintervallen behandelt wurden, wurde in einigen Studien über eine Stauung der Atemwege, einschließlich der Fälle einer Stauung der oberen Atemwege, einer verstopften Nase und einer Nasopharyngitis, berichtet.
Bei Patienten, die mit erythropoetischen Mitteln behandelt wurden, wurden thrombotische/vaskuläre Ereignisse wie Myokardischämie, Myokardinfarkt, zerebrovaskuläre Unfälle (Hirnblutung und Hirninfarkt), transitorische ischämische Attacken, tiefe Venenthrombose, arterielle Thrombose, Lungenembolie, Aneurysma, Retin beobachtet Gerinnung in der künstlichen Niere.
Nach monatelanger oder jahrelanger Behandlung mit Epoetin alfa wurde eine Antikörper-vermittelte Erythroblastopenie (PRCA) beobachtet. Bei den meisten dieser Patienten wurden Antikörper gegen Erythropoietin beobachtet (siehe Abschnitte 4.3 und 4.4).
Ausdruck von unerwünschten Ereignissen
In diesem Abschnitt werden die Häufigkeiten von Nebenwirkungen wie folgt definiert: Sehr häufig (> 1/10); häufig (> 1/100 bis 1/1 000 bis 1/10 000 a
Innerhalb jeder Häufigkeitsklasse werden die Nebenwirkungen in absteigender Reihenfolge ihres Schweregrades dargestellt.
Die Häufigkeit kann je nach Indikation variieren
Erwachsene und pädiatrische Hämodialysepatienten, erwachsene Patienten mit Peritonealdialyse und erwachsene Patienten mit Niereninsuffizienz, die noch nicht dialysepflichtig sind
Die häufigste Nebenwirkung im Rahmen einer Behandlung mit Epoetin alfa ist ein dosisabhängiger Blutdruckanstieg oder eine Verschlechterung einer vorbestehenden Hypertonie. Dieser Blutdruckanstieg kann pharmakologisch behandelt werden. Darüber hinaus wird eine Überwachung des Blutdrucks empfohlen zu Beginn der Therapie. In Einzelfällen sind bei Patienten mit normalem oder niedrigem Blutdruck auch folgende Reaktionen aufgetreten: hypertensive Krise mit Symptomen ähnlich einer Enzephalopathie (Kopfschmerzen und Verwirrtheitszustand) und generalisierte tonikoklonische Anfälle, die eine sofortige medizinische Intervention und intensive Behandlung erfordern zu plötzlichen akuten migräneähnlichen Kopfschmerzen, die ein Warnzeichen sein können.
Insbesondere bei Patienten mit Neigung zur Hypotonie oder mit Komplikationen arteriovenöser Fisteln (Stenosen, Aneurysmen etc.) kann es zu Shuntthrombosen kommen, bei denen eine frühzeitige Revision des Shunts und eine antithrombotische Prophylaxe, zum Beispiel mit Acetylsalicylsäure, empfohlen werden. .
Erwachsene Krebspatienten unter Chemotherapie mit symptomatischer Anämie
Bei Patienten, die mit Epoetin alfa behandelt werden, kann Bluthochdruck auftreten. Folglich sollte eine engmaschige Überwachung von Hämoglobin und Blutdruck durchgeführt werden.
Bei Patienten, die mit Erythropoetika behandelt wurden, wurde eine erhöhte Inzidenz vaskulärer thrombotischer Ereignisse beobachtet (siehe Abschnitt 4.4 und Abschnitt 4.8 – Allgemeine Hinweise).
Patientenkandidaten für eine Operation
Unabhängig von der Behandlung mit Erythropoietin können thromboembolische Ereignisse nach wiederholten Phlebotomien bei chirurgischen Patienten mit kardiovaskulärer Grunderkrankung auftreten.
Daher sollte bei solchen Patienten routinemäßig das entnommene Blutvolumen ersetzt werden. Bei Patienten mit Ausgangshämoglobin > 13 g/dl kann die Möglichkeit, dass die Behandlung mit Retacrit mit einem erhöhten Risiko für postoperative thrombotische/vaskuläre Ereignisse verbunden ist, nicht ausgeschlossen werden.
Meldung von vermuteten Nebenwirkungen
Die Meldung vermuteter Nebenwirkungen, die nach der Zulassung des Arzneimittels auftreten, ist wichtig, da sie eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem zu melden.
04.9 Überdosierung -
Das therapeutische Fenster von Erythropoietin ist sehr breit.Eine Überdosierung von Erythropoietin kann Wirkungen hervorrufen, die die pharmakologischen Wirkungen des Hormons erweitern. Bei zu hohen Hämoglobinwerten kann eine Phlebotomie durchgeführt werden. Gegebenenfalls sollte eine zusätzliche unterstützende Betreuung erfolgen.
05.0 PHARMAKOLOGISCHE EIGENSCHAFTEN -
05.1 "Pharmakodynamische Eigenschaften -
Pharmakotherapeutische Gruppe: Andere Antianämika, Erythropoietin
ATC-Code: B03XA01
Retacrit ist ein Biosimilar. Ausführliche Informationen finden Sie auf der Website der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu
Pharmakodynamische Wirkungen
Erythropoietin ist ein Glykoprotein, das als Mitose-stimulierender Faktor und Differenzierungshormon die Produktion von Erythrozyten aus den Vorläufern des Stammkompartiments stimuliert Das scheinbare Molekulargewicht von Erythropoietin beträgt 32.000-40.000 Dalton. Der Proteinanteil des Moleküls macht etwa 58 % seines Gesamtmolekulargewichts aus und besteht aus 165 Aminosäuren. Die vier Kohlenhydratketten sind durch drei N-glykosidische Bindungen und eine oglykosidische Bindung mit dem Protein verbunden. Hinsichtlich der Aminosäuresequenz und Kohlenhydratzusammensetzung ist Epoetin zeta identisch mit endogenem humanem Erythropoietin, das aus dem Urin anämischer Patienten isoliert wurde. Die biologische Wirksamkeit von Erythropoietin wurde in verschiedenen Tiermodellen nachgewiesen in vivo (normale und anämische Ratten, polyzythämische Mäuse). Nach Erythropoietin-Gabe steigen die Erythrozytenzahl, die Hämoglobinwerte und die Retikulozytenzahl zusammen mit der 59Fe-Einbaurate an. In Tests in vitro (Maus-Milzzellkultur) wurde nach Inkubation mit Erythropoietin ein erhöhter Einbau von 3H-Thymidin in kernhaltige erythroide Zellen der Milz beobachtet.
Durch Zellkulturen des menschlichen Knochenmarks wurde gezeigt, dass Erythropoietin spezifisch die Erythropoese stimuliert, ohne die Leukopoese zu verändern. Auf Knochenmarkzellen wurde keine zytotoxische Aktivität von Erythropoietin beobachtet.
Ähnlich wie bei anderen hämatopoetischen Wachstumsfaktoren wurde Erythropoietin nachgewiesen in vitro stimulierende Eigenschaften der menschlichen Endothelzellen besitzen.
Erwachsene Patienten mit Niereninsuffizienz, die noch keine Dialyse erhalten
In 2 Studien mit verlängerten Dosisintervallen zu Erythropoietin (3-mal pro Woche, einmal pro Woche, einmal alle 2 Wochen und einmal alle 4 Wochen) hielten einige Patienten mit längeren Dosisbereichen keine angemessenen Hämoglobinspiegel und die Protokollanforderungen für das Absetzen, die durch die Hämoglobin- Werte eingehalten werden (0 % bei einmal wöchentlicher Dosierung, 3,7 % bei einmal alle zwei Wochen verabreichter Dosierung und 3,3 % bei einmal alle 4 Wochen verabreichten Gruppen).
Klinische Wirksamkeit und Sicherheit
An drei placebokontrollierten Studien nahmen 721 Krebspatienten teil, die eine platinfreie Chemotherapie erhielten, davon 389 mit hämatologischen Malignomen (221 mit multiplem Myelom, 144 mit Non-Hodgkin-Lymphom und 24 mit anderen hämatologischen malignen Erkrankungen) und 332 mit soliden Tumoren (172 Mammatumoren, 64 gynäkologische , 23 Lungen-, 22 Prostata-, 21 Magen-Darm- und 30 andere). An zwei großen offenen Studien nahmen 2 697 Krebspatienten teil, die eine platinfreie Chemotherapie erhielten, darunter 1 895 mit soliden Tumoren (683 Brust-, 260 Lungen-, 174 Gynäkologie-, 300 Magen-Darm- und 478) und 802 mit hämatologischen Malignomen.
In einer prospektiven, randomisierten, doppelblinden, placebokontrollierten Studie, die an 375 anämischen Patienten mit verschiedenen nicht-myeloischen Neoplasien durchgeführt wurde und eine platinfreie Chemotherapie erhielten, wurde eine signifikante Verringerung der mit Anämie verbundenen Folgeerscheinungen (wie Müdigkeit, Asthenie und Reduktionsaktivität) ), gemessen mit den folgenden Bewertungsinstrumenten: der allgemeinen Bewertungsskala FACT-An (Functional Assessment of Cancer Therapy-Anemia), der Müdigkeitsbewertungsskala FACT-An und der Cancer Linear Analogue Scale (CLAS). Zwei weitere randomisierte, placebokontrollierte Studien mit weniger Patienten zeigten keine signifikante Verbesserung der Lebensqualitätsparameter, die mit der EORTC-QLQ-C30-Skala bzw. CLAS bewertet wurden.
Erythropoietin ist ein Wachstumsfaktor, der in erster Linie die Produktion von Erythrozyten stimuliert.Erythropoietin-Rezeptoren können auf der Oberfläche verschiedener Arten von Tumorzellen exprimiert werden.
Überleben und Tumorprogression wurden in fünf großen kontrollierten Studien analysiert, an denen insgesamt 2.833 Patienten teilnahmen, darunter vier doppelblinde, placebokontrollierte Studien und eine offene Studie. Diese Studien schlossen Patienten ein, die eine Chemotherapie erhielten (zwei Studien) oder Patientenpopulationen, bei denen keine Erythropoese-stimulierenden Mittel indiziert sind: Krebspatienten mit Anämie, die sich keiner Chemotherapie unterziehen, und Patienten mit Kopf-Hals-Tumoren, die sich einer Strahlentherapie unterziehen. In zwei Studien wurde das Zielhämoglobin Konzentration > 13 g/dL, in den restlichen Studien 12-14 g/dL. In der Open-Label-Studie zeigte sich kein Unterschied im Gesamtüberleben zwischen Patienten, die mit rekombinantem humanem Erythropoietin behandelt wurden, gegenüber Kontrollen. kontrollierten Studien, das Hazard Ratio (Gefahrenquote) für das Gesamtüberleben lag zwischen 1,25 und 2,47 zugunsten der Kontrollen. Im Vergleich zu Kontrollen beobachteten diese Studien einen statistisch signifikanten, konstanten und ungeklärten Anstieg der Mortalität bei Patienten mit Anämie, die mit mehreren häufigen malignen Erkrankungen einhergingen und mit rekombinantem humanem Erythropoietin behandelt wurden. Die Ergebnisse des Gesamtüberlebens der Studien konnten durch die Unterschiede in der Häufigkeit von Thrombosen und damit verbundenen Komplikationen bei mit rekombinantem humanem Erythropoietin behandelten Patienten und bei Kontrollpersonen nicht zufriedenstellend erklärt werden.
Darüber hinaus wurde eine systematische Überprüfung von über 9.000 Krebspatienten durchgeführt, die an 57 klinischen Studien teilnahmen. Die Metaanalyse der Gesamtüberlebensdaten ergab eine geschätzte Hazard Ratio von 1,08 zugunsten der Kontrollen (95%-KI: 0,99; 1,18; 42 Studien und 8 167 Patienten). Bei Patienten, die mit rekombinantem humanem Erythropoietin behandelt wurden, wurde ein erhöhtes relatives Risiko für thromboembolische Ereignisse beobachtet (RR 1,67, 95 %-KI: 1,35, 2,06, 35 Studien und 6 769 Patienten). Bei Krebspatienten, die mit rekombinantem humanem Erythropoietin behandelt werden, besteht ein erhöhtes Risiko für thromboembolische Ereignisse, und eine negative Auswirkung auf das Gesamtüberleben kann nicht ausgeschlossen werden. Es ist nicht bekannt, inwieweit diese Daten auf die Verabreichung von rekombinantem humanem Erythropoietin an Krebspatienten zurückzuführen sind, die eine Chemotherapie erhalten, um Hämoglobinkonzentrationen unter 13 g/dl zu erreichen, da nur wenige Patienten mit den beschriebenen Merkmalen in die überprüften Daten eingeschlossen wurden.
Eine Einzelpatientendatenanalyse wurde auch bei über 13.900 Krebspatienten (Chemo-Radio-, Chemoradium- oder keine Therapie) durchgeführt, die an 53 kontrollierten klinischen Studien mit verschiedenen Epoetinen teilnahmen Kontrollen (95 %-KI: 1,00; 1,12: 53 Studien und 13 933 Patienten) und für Krebspatienten, die eine Chemotherapie erhielten, betrug die Gesamtüberlebens-Hazard-Ratio 1,04 (95 %-KI: 0,97; 1,11; 38 Studien und 10 441 Patienten). -Analyse belegt auch ein konsistentes und signifikant erhöhtes relatives Risiko für thromboembolische Ereignisse bei Krebspatienten, die mit rekombinantem humanem Erythropoietin behandelt werden (siehe Abschnitt 4.4).
In einer randomisierten, doppelblinden, placebokontrollierten Studie mit 4.038 nicht dialysierten CNI-Patienten mit Typ-2-Diabetes und Hämoglobinwerten ≤ 11 g/dl wurden die Patienten entweder mit Darbepoetin alfa behandelt, um Hämoglobinwerte von 13 g/dl zu erreichen oder Placebo (siehe Abschnitt 4.4). Die Studie erreichte keines der primären Ziele hinsichtlich der Verringerung des Risikos der damit verbundenen Mortalität, der kardiovaskulären Morbidität und der Entwicklung einer Nierenerkrankung im Endstadium (ESRD).Analysen der einzelnen Komponenten der zusammengesetzten Endpunkte zeigten eine HR (95 KI: Tod 1,05 (0,92; 1,21), Schlaganfall 1,92 (1,38; 2,68), kongestive Herzinsuffizienz (CHF) 0,89 (0,74; 1,08), Myokardinfarkt (MI) 0,96 (0,75; 1,23), Krankenhausaufenthalt wegen Myokardischämie 0,84 ( 0,55, 1,27), ESRD 1,02 (0,87, 1,18).
Es wurden gepoolte Analysen von Post-hoc-Daten aus klinischen Studien mit ESA durchgeführt, die bei Patienten mit CNI (unter Dialyse, nicht unter Dialyse, mit oder ohne Diabetes) durchgeführt wurden. Es zeigte sich ein Trend zu steigenden Risikoschätzungen für die Gesamtmortalität sowie für kardiovaskuläre und zerebrovaskuläre Ereignisse im Zusammenhang mit den höchsten kumulativen ESA-Dosen, unabhängig vom Diabetes- oder Dialysestatus (siehe Abschnitte 4.2 und 4.4).
05.2 "Pharmakokinetische Eigenschaften -
Intravenöser Verabreichungsweg
Die Messung von Erythropoietin nach wiederholter intravenöser Verabreichung zeigte eine „Halbwertszeit von ca. 4 Stunden bei gesunden Probanden und eine“ etwas längere Halbwertszeit bei Patienten mit Niereninsuffizienz (ca. 5 Stunden). Bei Kindern wurde eine Halbwertszeit von etwa 6 Stunden berichtet.
Art der subkutanen Verabreichung
Nach subkutaner Injektion sind die Serum-Erythropoietin-Spiegel viel niedriger als die intravenösen, steigen langsam an und erreichen ihren Höhepunkt zwischen 12 und 18 Stunden nach der Verabreichung. Dieser Peak liegt immer deutlich unter dem, der intravenös erreicht wird (ca. 1/20).
Es gibt keine Kumulationsphänomene: Die Konzentrationen bleiben gleich, egal ob sie 24 Stunden nach der ersten Injektion oder 24 Stunden nach der letzten Injektion festgestellt werden.
Die Halbwertszeit bei subkutaner Verabreichung ist schwer zu beurteilen und wird auf ca. 24 Stunden geschätzt.Die Bioverfügbarkeit von subkutan injizierbarem Erythropoietin ist viel geringer als die des intravenösen Arzneimittels: ca. 20 %.
05.3 Präklinische Daten zur Sicherheit -
In einigen präklinischen Toxikologiestudien an Hunden und Ratten, jedoch nicht an Affen, wurde eine Erythropoietin-Therapie mit einer subklinischen Knochenmarkfibrose in Verbindung gebracht (Knochenmarksfibrose ist eine bekannte Komplikation des chronischen Nierenversagens beim Menschen und kann mit sekundärem Hyperparathyreoidismus oder unbekannten Faktoren zusammenhängen). . In einer Studie mit Hämodialysepatienten, die 3 Jahre lang mit Erythropoietin behandelt wurden, war die Inzidenz von Knochenmarkfibrose im Vergleich zu einer entsprechenden Gruppe von Kontrollpatienten unter Dialyse, die nicht mit Erythropoietin behandelt wurden, nicht erhöht.
Tierexperimentelle Studien haben gezeigt, dass Erythropoietin das Körpergewicht des Fötus verringert, den Ossifikationsprozess verzögert und die fötale Sterblichkeit erhöht, wenn es in wöchentlichen Dosen gegeben wird, die etwa das 20-fache der für den Menschen empfohlenen Dosis betragen. Diese Veränderungen werden als sekundär zu der reduzierten Zunahme des mütterlichen Körpergewichts interpretiert.
Erythropoietin zeigte in Mutagenitätstests an Bakterien- und Säugetierzellkulturen keine Aktivität und in vivo in einem Maus-Mikronukleus-Test. Langzeitstudien zur Kanzerogenität wurden nicht durchgeführt. In der Literatur gibt es widersprüchliche Daten bezüglich der Möglichkeit, dass Erythropoietin eine wichtige Rolle bei der Proliferation von Krebszellen spielt. Diese Daten basieren auf erhaltenen Ergebnissen in vitro aus menschlichen Tumorgewebeproben; ihr Anwendungsbereich im klinischen Umfeld ist jedoch unklar.
06.0 PHARMAZEUTISCHE INFORMATIONEN -
06.1 Hilfsstoffe -
Dinatriumphosphat-Dihydrat
Monobasisches Natriumphosphat-Dihydrat
Natriumchlorid
Calciumchlorid-Dihydrat
Polysorbat 20
Glycin
Leucin
Isoleucin
Threonin
Glutaminsäure
Phenylalanin
Wasser für Injektionslösungen
Natriumhydroxid (um den pH-Wert einzustellen)
Salzsäure (zur Einstellung des pH-Wertes)
06.2 Inkompatibilität "-
Da keine Inkompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
06.3 Gültigkeitsdauer "-
30 Monate
06.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung -
Im Kühlschrank lagern (2 ° C - 8 ° C). Nicht einfrieren.
Bewahren Sie die Fertigspritze im Umkarton auf, um das Arzneimittel vor Licht zu schützen.
Während der ambulanten Anwendung kann der Patient das Produkt aus dem Kühlschrank nehmen und bei Raumtemperatur (nicht über 25 ° C) einmalig für maximal 3 Tage lagern.
06.5 Art der unmittelbaren Verpackung und Inhalt des Packstücks -
0,3 ml Lösung in einer Fertigspritze aus Glas Typ I mit fester Stahlnadel und PTFE-beschichtetem Kolbenstopfen mit oder ohne Nadelschutz.
Eine Packung enthält 1 oder 6 Fertigspritzen.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
06.6 Gebrauchs- und Handhabungshinweise -
Hinweise zum Umgang mit Retacrit:
1. Nachdem Sie eine Spritze aus der Blisterpackung entnommen haben, prüfen Sie, ob die Lösung klar, farblos und praktisch frei von sichtbaren Partikeln ist.
2. Entfernen Sie die Nadelschutzkappe und drücken Sie die Luft aus der Nadel und der Spritze, indem Sie die Spritze aufrecht halten und den Kolben vorsichtig nach oben drücken.
3. Die Spritze ist gebrauchsfertig.
Retacrit sollte nicht angewendet werden, wenn einer der folgenden Fälle eintritt:
• die Blisterpackung geöffnet oder anderweitig beschädigt ist;
• die Lösung nicht farblos ist oder sichtbare Schwebeteilchen enthält;
• c „Flüssigkeit ist aus der Fertigspritze ausgetreten oder es ist Kondenswasser in der noch versiegelten Blisterpackung sichtbar;
• das Arzneimittel versehentlich eingefroren wurde.
Dieses Arzneimittel ist nur zur einmaligen Anwendung bestimmt.
Nicht schütteln.
Nicht verwendete Arzneimittel und Abfälle aus diesem Arzneimittel müssen gemäß den örtlichen Vorschriften entsorgt werden.
07.0 INHABER DER "MARKETING GENEHMIGUNG" -
Hospira UK Limited
Horizont
Honigstraße
Hurley
Jungfrauenkopf
SL6 6RJ
Vereinigtes Königreich
08.0 NUMMER DER VERMARKTUNGSBERECHTIGUNG -
EU / 1/07/431/001 Fertigspritze
EU / 1/07/431/002 Fertigspritze
EU / 1/07/431/026 Fertigspritze mit Nadelschutz
EU / 1/07/431/027 Fertigspritze mit Nadelschutz
038381012
038381024
038381265
038381277
09.0 DATUM DER ERSTEN GENEHMIGUNG ODER ERNEUERUNG DER GENEHMIGUNG -
Datum der Erstzulassung: 18. Dezember 2007
Datum der letzten Verlängerung: 15. November 2012
10.0 DATUM DER ÜBERARBEITUNG DES TEXTs -
D.CCE September 2016
11.0 FÜR RADIOPARMAZEUGIEN VOLLSTÄNDIGE DATEN ZUR INTERNEN STRAHLENDOSIMETRIE -
12.0 FÜR FUNKDROGEN, WEITERE DETAILLIERTE ANWEISUNGEN ZUR EXTEMPORÄREN ZUBEREITUNG UND QUALITÄTSKONTROLLE -