Wirkstoffe: Strontium (Strontiumranelat)
OSSEOR 2 g Granulat zur Herstellung einer Suspension zum Einnehmen
Indikationen Warum wird Osseor verwendet? Wofür ist das?
OSSEOR ist ein Arzneimittel zur Behandlung schwerer Osteoporose:
- bei postmenopausalen Frauen,
- bei erwachsenen Männern,
bei hohem Frakturrisiko, bei dem auf alternative Behandlungsmethoden nicht zurückgegriffen werden kann. Bei postmenopausalen Frauen reduziert Strontiumranelat das Risiko von Wirbelsäulen- und Hüftfrakturen.
Osteoporose
Der Körper zerstört ständig alten Knochen und bildet neuen Knochen.Bei Osteoporose zerstört der Körper mehr Knochen als gebildet wird, so dass allmählich Knochenschwund auftritt und die Knochen dünner und brüchiger werden.Es tritt insbesondere bei Frauen nach den Wechseljahren auf.
Viele Menschen mit Osteoporose haben keine Symptome und es ist möglich, nicht einmal zu wissen, dass Sie Osteoporose haben. Osteoporose prädisponiert jedoch für Frakturen (Knochenbrüche) insbesondere in der Wirbelsäule, Hüfte und Handgelenken.
So funktioniert OSSEOR
OSSEOR, das den Wirkstoff Strontiumranelat enthält, gehört zu einer Gruppe von Arzneimitteln zur Behandlung von Knochenerkrankungen.
OSSEOR reduziert die Knochenzerstörung und stimuliert den Knochenaufbau, wodurch das Risiko von Frakturen reduziert wird. Der neu gebildete Knochen ist von normaler Qualität.
Kontraindikationen Wenn Osseor nicht verwendet werden sollte
Nehmen Sie OSSEOR® nicht ein
- wenn Sie allergisch gegen Strontiumranelat oder einen der in Abschnitt 6. genannten sonstigen Bestandteile von OSSEOR sind.
- wenn Sie eine Thrombose haben oder jemals hatten (z. B. die die Blutgefäße im Bein oder in der Lunge betrifft).
- wenn Sie dauerhaft oder für eine bestimmte Zeit immobilisiert sind, z. B. wenn Sie im Rollstuhl sitzen, bettlägerig sind oder sich einer Operation unterziehen müssen oder sich in der postoperativen Genesung befinden. Das Risiko einer Venenthrombose (Thrombose im Bein oder in der Lunge) kann bei längerer Ruhigstellung höher sein.
- wenn Sie eine bekannte ischämische Herzerkrankung oder eine zerebrovaskuläre Erkrankung haben, z. wenn bei Ihnen ein Herzinfarkt, Schlaganfall oder eine vorübergehende ischämische Attacke (vorübergehende Verringerung der Durchblutung des Gehirns; auch bekannt als „Mini-Schlaganfall“), Angina pectoris oder eine Blockade der Blutgefäße im Herzen oder Gehirn diagnostiziert wurde .
- wenn Sie Probleme mit Ihrer Durchblutung haben oder hatten (periphere arterielle Verschlusskrankheit) oder wenn Sie an den Arterien in Ihren Beinen operiert wurden.
- wenn Sie hohen Blutdruck haben, der durch die Behandlung nicht kontrolliert werden kann.
Vorsichtsmaßnahmen für die Anwendung Was sollten Sie vor der Einnahme von Osseor® beachten?
Bitte sprechen Sie mit Ihrem Arzt oder Apotheker, bevor Sie OSSEOR einnehmen:
- wenn bei Ihnen das Risiko einer Herzerkrankung besteht; dazu gehören Bluthochdruck, hoher Cholesterinspiegel, Diabetes, Rauchen – wenn Sie ein Thromboserisiko haben
- wenn Sie eine schwere Nierenerkrankung haben.
Während der Behandlung mit OSSEOR wird Ihr Arzt den Zustand Ihres Herzens und Ihrer Blutgefäße in regelmäßigen Abständen, normalerweise alle 6 bis 12 Monate, untersuchen.
Wenn während der Behandlung eine allergische Reaktion auftritt (wie Anschwellen von Gesicht, Zunge oder Rachen, Atem- oder Schluckbeschwerden, Hautausschlag), sollten Sie die Einnahme von OSSEOR sofort abbrechen und Ihren Arzt aufsuchen (siehe Abschnitt 4).
Bei der Anwendung von OSSEOR wurde über potenziell lebensbedrohliche Hautausschläge (Stevens-Johnson-Syndrom (SJS), toxische epidermale Nekrolyse und schwere Überempfindlichkeitsreaktionen (DRESS)) berichtet.
Das größte Risiko für das Auftreten schwerer Hautreaktionen besteht in den ersten Behandlungswochen des Stevens-Johnson-Syndroms und der toxischen epidermalen Nekrolyse und im Allgemeinen etwa 3-6 Wochen bei DRESS.
Wenn bei Ihnen ein Hautausschlag oder schwerwiegende Hautsymptome auftreten (siehe Abschnitt 4), beenden Sie die Einnahme von OSSEOR, wenden Sie sich sofort an Ihren Arzt und teilen Sie mit, dass Sie dieses Arzneimittel einnehmen.
Wenn bei Ihnen während der Anwendung von OSSEOR ein Stevens-Johnson-Syndrom, eine toxische epidermale Nekrolyse oder DRESS aufgetreten ist, sollten Sie die Behandlung mit OSSEOR nie wieder aufnehmen.
Wenn Sie asiatischer Abstammung sind, sprechen Sie mit Ihrem Arzt, bevor Sie OSSEOR einnehmen, da Sie möglicherweise ein höheres Risiko für Hautreaktionen haben.
Kinder und Jugendliche
OSSEOR ist nicht zur Anwendung bei Kindern und Jugendlichen (unter 18 Jahren) bestimmt.
Wechselwirkungen Welche Medikamente oder Lebensmittel können die Wirkung von Osseor® verändern?
Einnahme von OSSEOR® zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen, kürzlich andere Arzneimittel eingenommen haben oder beabsichtigen andere Arzneimittel einzunehmen.
Brechen Sie die Einnahme von OSSEOR ab, wenn Sie orale Tetracycline wie Doxycyclin oder Chinolone wie Ciprofloxacin (zwei Arten von Antibiotika) einnehmen müssen. Sie können OSSEOR wieder aufnehmen, wenn Sie die Einnahme dieser Antibiotika beendet haben. Wenn Sie sich nicht sicher sind, fragen Sie Ihren Arzt oder Apotheker Wenn Sie kalziumhaltige Arzneimittel einnehmen, lassen Sie vor der Einnahme von OSSEOR mindestens 2 Stunden verstreichen.Wenn Sie Antazida (Arzneimittel gegen Sodbrennen) einnehmen, nehmen Sie diese mindestens 2 Stunden nach der Einnahme von OSSEOR ein. Ist dies nicht möglich, ist die gleichzeitige Einnahme beider Medikamente akzeptabel.
Wenn es notwendig ist, den Kalziumspiegel im Blut oder Urin zu testen, müssen Sie das Labor darüber informieren, dass Sie OSSEOR einnehmen, da es einige Testmethoden beeinträchtigen kann.
Einnahme von OSSEOR zusammen mit Speisen und Getränken
Nahrung, Milch und seine Derivate reduzieren die Aufnahme von Strontiumranelat.Es wird empfohlen, OSSEOR in den Pausen zwischen den Mahlzeiten einzunehmen, vorzugsweise vor dem Schlafengehen, mindestens zwei Stunden nach der Einnahme von Nahrung, Milch und Milchderivaten oder Kalziumpräparaten.
Warnungen Es ist wichtig zu wissen, dass:
Schwangerschaft und Stillzeit
Nehmen Sie OSSEOR nicht während der Schwangerschaft oder Stillzeit ein. Im Falle einer versehentlichen Einnahme während der Schwangerschaft oder Stillzeit beenden Sie die Einnahme des Arzneimittels sofort und informieren Sie Ihren Arzt.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Es ist unwahrscheinlich, dass OSSEOR die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen beeinflusst.
OSSEOR enthält Aspartam (E951):
Wenn Sie Phenylketonurie (eine seltene erbliche Stoffwechselstörung) haben, konsultieren Sie Ihren Arzt, bevor Sie mit der Einnahme dieses Arzneimittels beginnen.
Dosis, Methode und Zeitpunkt der Verabreichung Wie ist Osseor anzuwenden: Dosierung
Die Behandlung sollte nur von einem in der Behandlung von Osteoporose erfahrenen Arzt eingeleitet werden.
Nehmen Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt oder Apotheker ein. Fragen Sie im Zweifelsfall Ihren Arzt oder Apotheker.
OSSEOR ist zur oralen Anwendung bestimmt. Die empfohlene Dosis beträgt einen Beutel zu 2 g pro Tag.
Es wird empfohlen, OSSEOR vor dem Schlafengehen einzunehmen, vorzugsweise mindestens 2 Stunden nach dem Abendessen. Wenn Sie möchten, können Sie nach der Einnahme von OSSEOR auch sofort zu Bett gehen.
Nehmen Sie das in den Beuteln enthaltene Granulat, nachdem Sie es in einem Glas suspendiert haben, das mindestens 30 ml Wasser enthält (etwa ein Drittel eines Standardglases) (siehe nachstehende Anweisungen). OSSEOR kann mit Milch und ihren Derivaten interagieren, daher ist es Es ist wichtig, dass OSSEOR nur mit Wasser gemischt wird, um sicherzustellen, dass das Arzneimittel richtig wirkt.
- Gießen Sie das Granulat aus dem Beutel in ein Glas;
- Wasser hinzufügen;
- Rühren, bis das Granulat vollständig im Wasser dispergiert ist.
Sofort trinken. Warten Sie nicht länger als 24 Stunden, bevor Sie die Suspension trinken. Wenn Sie das Arzneimittel aus irgendeinem Grund nicht sofort einnehmen können, denken Sie daran, es vor dem Trinken noch einmal zu mischen.
Ihr Arzt wird Ihnen möglicherweise raten, zusätzlich zu OSSEOR Calcium- und Vitamin-D-Präparate einzunehmen. Nehmen Sie keine Kalziumpräparate vor dem Schlafengehen gleichzeitig mit OSSEOR ein.
Ihr Arzt wird Ihnen sagen, wie lange Sie OSSEOR einnehmen sollen. Die Behandlung von Osteoporose dauert in der Regel lange. Es ist wichtig, dass Sie OSSEOR so lange einnehmen, wie Ihr Arzt es Ihnen verordnet.
Überdosierung Was ist zu tun, wenn Sie zu viel Osseor eingenommen haben?
Wenn Sie eine größere Menge von OSSEOR eingenommen haben, als Sie sollten
Wenn Sie eine größere Menge von OSSEOR-Beutel eingenommen haben, als Ihr Arzt verordnet hat, informieren Sie bitte Ihren Arzt oder Apotheker. Sie können Ihnen raten, Milch zu trinken oder Antazida einzunehmen, um die Aufnahme des Wirkstoffs zu verringern.
Wenn Sie die Einnahme von OSSEOR® vergessen haben
Nehmen Sie nicht die doppelte Dosis ein, wenn Sie die vorherige Einnahme vergessen haben. Nehmen Sie einfach die nächste Dosis zum vereinbarten Zeitpunkt ein.
Wenn Sie die Einnahme von OSSEOR® abbrechen
Es ist wichtig, dass Sie OSSEOR so lange einnehmen, wie Ihr Arzt es Ihnen verschrieben hat.OSSEOR kann schwere Osteoporose nur behandeln, wenn es kontinuierlich eingenommen wird.Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Nebenwirkungen Was sind die Nebenwirkungen von Osseor
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Brechen Sie die Einnahme von OSSEOR ab und informieren Sie sofort Ihren Arzt, wenn eine der folgenden Nebenwirkungen auftritt:
Häufig (kann bis zu 1 von 10 Patienten betreffen):
- Herzinfarkt: Plötzliche, zusammendrückende Schmerzen in der Brust, die sich auf den linken Arm, Kiefer, Bauch, Rücken und / oder Schultern erstrecken können. Andere Symptome können sein: Übelkeit/Erbrechen, Schwitzen, Kurzatmigkeit, Herzklopfen, (extreme) Müdigkeit und/oder Schwindel. Bei Patienten mit hohem Risiko für Herzerkrankungen kann ein Herzinfarkt mit häufiger Häufigkeit auftreten. Wenn Sie ein Hochrisikopatient sind, wird Ihr Arzt OSSEOR nicht verschreiben.
- Blutgerinnsel in den Venen (Thrombose): Schmerzen, Rötung, Anschwellen der Beine, plötzliche Brustschmerzen oder Atembeschwerden.
Selten (kann bis zu 1 von 1.000 Patienten betreffen):
- Anzeichen einer schweren Überempfindlichkeitsreaktion (DRESS): zunächst grippeähnliche Symptome und Hautausschlag, dann ausgedehnter Hautausschlag mit hohem Fieber (gelegentlich), erhöhte Leberenzymwerte bei Blutuntersuchungen (gelegentlich), Anstieg einer bestimmten Art von weißem Blutkörperchen (Eosinophilie) (selten) und vergrößerte Lymphknoten (gelegentlich).
Sehr selten (kann bis zu 1 von 10.000 Behandelten betreffen):
- Anzeichen eines potenziell lebensbedrohlichen Hautausschlags (Stevens-Johnson-Syndrom, toxische epidermale Nekrolyse): Anfänglich als rötliche zielartige Flecken oder kreisrunde Flecken, oft mit zentralen Blasen am Rumpf. Zusätzliche Anzeichen können Geschwüre in Mund, Rachen, Nase, Genitalien und Konjunktivitis (geschwollene und rote Augen) sein. Diese potenziell lebensbedrohlichen Hautausschläge werden oft von grippeähnlichen Symptomen begleitet. Der Ausschlag kann zu Blasenbildung am ganzen Körper oder zum Abschälen der Haut führen.
Andere mögliche Nebenwirkungen
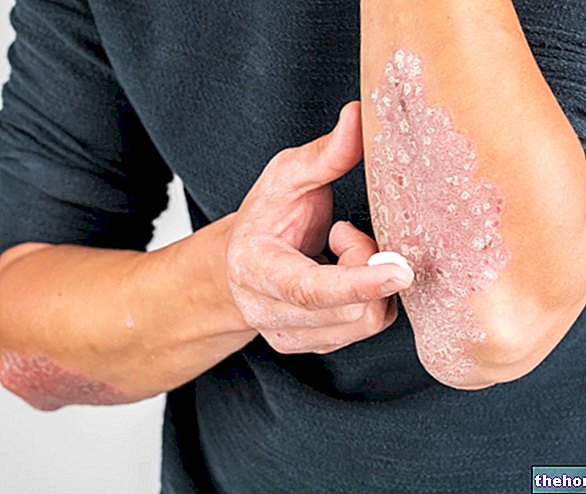
Sehr häufig (kann mehr als 1 von 10 Behandelten betreffen): Juckreiz, Nesselsucht, Hautausschlag, Angioödem (wie Schwellung von Gesicht, Zunge oder Rachen, Atem- oder Schluckbeschwerden), Schmerzen in Knochen, Gliedmaßen, Muskeln und/oder in den Gelenken, Muskelkrämpfe.
Häufig: Erbrechen, Bauchschmerzen, Reflux, Verdauungsstörungen, Verstopfung, Blähungen, Schlafstörungen, Leberentzündung (Hepatitis), Anschwellen der Gliedmaßen, bronchiale Hyperreaktivität (Symptome sind pfeifende Atmung, Kurzatmigkeit und Husten), erhöhtes Muskelniveau Enzym (Kreatinphosphokinase). Übelkeit, Durchfall, Kopfschmerzen, Ekzeme, Gedächtnisstörungen, Ohnmacht, Kribbeln, Schwindel, Schwindel. Diese Nebenwirkungen sind jedoch leicht und vorübergehend und erfordern in der Regel kein Absetzen der Behandlung. Informieren Sie Ihren Arzt, wenn eine dieser Nebenwirkungen unangenehm oder anhaltend wird.
Gelegentlich (kann bis zu 1 von 100 Behandelten betreffen): Krämpfe, Reizung der Mundschleimhaut (wie Geschwüre im Mund und Zahnfleischentzündung), Haarausfall, Verwirrtheit, Übelkeit, Mundtrockenheit, Hautreizung.
Selten: Verminderte Produktion von Blutzellen im Knochenmark. Wenn Sie die Therapie aufgrund von Überempfindlichkeitsreaktionen abgebrochen haben, sollten Sie OSSEOR nicht erneut starten.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker, einschließlich aller möglichen Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem melden. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
Ablauf und Aufbewahrung
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Verwenden Sie dieses Arzneimittel nicht nach dem Verfallsdatum, das auf der Packung und dem Beutel nach dem Wort "Verwendbar" angegeben ist. Das Verfallsdatum bezieht sich auf den letzten Tag dieses Monats. Nach der Rekonstitution in Wasser ist die Suspension 24 Stunden haltbar.
Es wird jedoch empfohlen, die Suspension unmittelbar nach der Zubereitung zu trinken (siehe Abschnitt 3). Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Werfen Sie Arzneimittel nicht in das Abwasser oder den Hausmüll. Fragen Sie Ihren Apotheker, wie Sie Arzneimittel, die Sie nicht mehr verwenden, entsorgen. Dies trägt zum Schutz der Umwelt bei.
Zusammensetzung und Darreichungsform
Was OSSEOR enthält
- Der Wirkstoff ist Strontiumranelat. Jeder Beutel enthält 2 g Strontiumranelat.
- Die sonstigen Bestandteile sind Aspartam (E 951), Maltodextrin, Mannit (E 421).
Beschreibung wie OSSEOR aussieht und Inhalt der Packung
OSSEOR ist in Beuteln mit einem gelben Granulat zur Herstellung einer Suspension zum Einnehmen erhältlich. OSSEOR wird in Packungen mit 7, 14, 28, 56, 84 oder 100 Beuteln geliefert. Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
Quelle Packungsbeilage: AIFA (Italienische Arzneimittelbehörde). Im Januar 2016 veröffentlichter Inhalt. Die vorliegenden Informationen können nicht aktuell sein.
Um Zugriff auf die aktuellste Version zu erhalten, ist es ratsam, auf die Website der AIFA (Italienische Arzneimittelbehörde) zuzugreifen. Haftungsausschluss und nützliche Informationen.
01.0 BEZEICHNUNG DES ARZNEIMITTELS
OSSEOR 2G GRANULAT ZUR ORALEN SUSPENSION
▼ Arzneimittel, das einer zusätzlichen Überwachung unterliegt. Dies ermöglicht die schnelle Identifizierung neuer Sicherheitsinformationen. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Siehe Abschnitt 4.8 für Informationen zur Meldung von Nebenwirkungen.
02.0 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Jeder Beutel enthält 2 g Strontiumranelat.
Sonstiger Bestandteil mit bekannter Wirkung:
jeder Beutel enthält außerdem 20 mg Aspartam (E 951)
Die vollständige Auflistung der sonstigen Bestandteile finden Sie in Abschnitt 6.1.
03.0 DARREICHUNGSFORM
Granulat zur Herstellung einer Suspension zum Einnehmen
Gelbes Granulat
04.0 KLINISCHE INFORMATIONEN
04.1 Anwendungsgebiete
Behandlung schwerer Osteoporose:
• bei postmenopausalen Frauen,
• bei erwachsenen Männern,
bei hohem Frakturrisiko, bei denen eine Behandlung mit anderen zur Behandlung der Osteoporose zugelassenen Arzneimitteln beispielsweise aufgrund von Kontraindikationen oder Unverträglichkeiten nicht möglich ist.
Bei postmenopausalen Frauen reduziert Strontiumranelat das Risiko von Wirbel- und Hüftfrakturen (siehe Abschnitt 5.1).
Die Entscheidung, Strontiumranelat zu verschreiben, sollte auf einer Bewertung der Gesamtrisiken des einzelnen Patienten beruhen (siehe Abschnitte 4.3 und 4.4).
04.2 Dosierung und Art der Anwendung
Die Behandlung sollte nur von einem in der Behandlung von Osteoporose erfahrenen Arzt eingeleitet werden.
Dosierung
Die empfohlene Dosis beträgt einmal täglich einen Beutel zu 2 g zur oralen Verabreichung.
Aufgrund der Art der zu behandelnden Erkrankung ist Strontiumranelat für die Langzeitanwendung vorgesehen.
Die Resorption von Strontiumranelat aus der Nahrung, Milch und seinen Derivaten wird reduziert und daher sollte OSSEOR zwischen den Mahlzeiten eingenommen werden. Aufgrund seiner langsamen Resorption sollte OSSEOR vor dem Zubettgehen eingenommen werden, vorzugsweise mindestens zwei Stunden nach einer Mahlzeit (siehe Abschnitte 4.5 und 5.2).
Patienten, die mit Strontiumranelat behandelt werden, sollten Vitamin D- und Kalziumpräparate einnehmen, wenn ihre Nahrungsaufnahme nicht ausreicht.
Ältere Patienten
Die Wirksamkeit und Sicherheit von Strontiumranelat wurde in einer großen Stichprobe von erwachsenen Männern und postmenopausalen Frauen jeden Alters (bis zu 100 Jahre bei Aufnahme) mit Osteoporose nachgewiesen. Eine altersabhängige Dosisanpassung ist nicht erforderlich.
Patienten mit Niereninsuffizienz
Strontiumranelat wird bei Patienten mit schwerer Niereninsuffizienz (Kreatinin-Clearance unter 30 ml/min) nicht empfohlen (siehe Abschnitte 4.4 und 5.2). Bei Patienten mit leichter bis mittelschwerer Niereninsuffizienz (Kreatinin-Clearance 30 – 70 ml .) ist keine Dosisanpassung erforderlich / min) (siehe Abschnitte 4.4 und 5.2).
Patienten mit Leberinsuffizienz
Bei Patienten mit Leberinsuffizienz ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von OSSEOR bei Kindern unter 18 Jahren sind nicht erwiesen.Es liegen keine Daten vor.
Art der Verabreichung
Zur oralen Anwendung
Das Granulat der Beutel sollte nach Suspension in einem Glas mit mindestens 30 ml Wasser (etwa ein Drittel eines normalen Glases) eingenommen werden.
Obwohl Anwendungsstudien gezeigt haben, dass Strontiumranelat 24 Stunden nach der Zubereitung in Suspension stabil bleibt, sollte die Suspension sofort nach der Zubereitung getrunken werden.
04.3 Kontraindikationen
• Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
• Aktuelle oder frühere venöse Thromboembolien (VTE), einschließlich tiefer Venenthrombose und Lungenembolie.
• Vorübergehende oder dauerhafte Immobilisierung, beispielsweise aufgrund einer Operation oder eines längeren Bettaufenthalts.
• Festgestellte, aktuelle oder frühere ischämische Herzkrankheit, periphere arterielle Verschlusskrankheit und/oder zerebrovaskuläre Erkrankung.
• Unkontrollierter Bluthochdruck.
04.4 Besondere Warnhinweise und geeignete Vorsichtsmaßnahmen für die Anwendung
Ischämische kardiale Ereignisse
In einer gepoolten Analyse von placebokontrollierten randomisierten klinischen Studien bei postmenopausalen osteoporotischen Patienten wurde bei Patienten, die mit OSSEOR behandelt wurden, ein signifikanter Anstieg von Myokardinfarkten im Vergleich zu denen, die mit Placebo behandelt wurden, beobachtet (siehe Abschnitt 4.8).
Patienten sollten vor Beginn der Behandlung auf kardiovaskuläre Risiken untersucht werden.
Patienten mit signifikanten Risikofaktoren für kardiovaskuläre Ereignisse (z. B. Hypertonie, Hyperlipidämie, Diabetes mellitus, Rauchen) sollten nur nach sorgfältiger Abwägung mit Strontiumranelat behandelt werden (siehe Abschnitte 4.3 und 4.8).
Während der Behandlung mit OSSEOR sollten diese kardiovaskulären Risiken in regelmäßigen Abständen, in der Regel alle 6-12 Monate, überwacht werden.
Die Behandlung sollte abgebrochen werden, wenn der Patient eine ischämische Herzerkrankung, eine periphere arterielle Verschlusskrankheit, eine zerebrovaskuläre Erkrankung entwickelt oder wenn der Bluthochdruck nicht unter Kontrolle ist (siehe Abschnitt 4.3).
Venöse Thromboembolie
In placebokontrollierten Phase-III-Studien war die Behandlung mit Strontiumranelat mit einer erhöhten jährlichen Inzidenz von venösen Thromboembolien (VTE), einschließlich Lungenembolien, verbunden (siehe Abschnitt 4.8). Die Ursache für diesen Anstieg ist unbekannt. OSSEOR ist bei Patienten mit früherer venöser Thromboembolie kontraindiziert (siehe Abschnitt 4.3) und sollte bei Patienten mit VTE-Risiko mit Vorsicht angewendet werden.
Während der Behandlung von Patienten über 80 Jahren mit VTE-Risiko sollte die Notwendigkeit einer Fortsetzung der Behandlung mit OSSEOR neu bewertet werden. Im Falle einer Krankheit oder eines Zustands, der zur Ruhigstellung führt (siehe Abschnitt 4.3), sollte die Behandlung mit OSSEOR so schnell wie möglich abgebrochen und geeignete Präventivmaßnahmen ergriffen werden.Die Therapie sollte erst wieder aufgenommen werden, wenn der Zustand, der zur Ruhigstellung geführt hat, nicht abgeklungen ist und der Patient ist vollständig mobil. Wenn eine VTE auftritt, sollte OSSEOR abgesetzt werden.
Anwendung bei Patienten mit Niereninsuffizienz
Da bei Patienten mit schwerer Niereninsuffizienz, die Strontiumranelat erhalten, keine Daten zur Knochensicherheit vorliegen, wird OSSEOR bei Patienten mit einer Kreatinin-Clearance von weniger als 30 ml/min nicht empfohlen. (siehe Abschnitt 5.2). In Übereinstimmung mit der guten klinischen Praxis wird bei Patienten mit chronischem Nierenversagen eine regelmäßige Überwachung der Nierenfunktion empfohlen. Die Fortsetzung der OSSEOR-Therapie bei Patienten, die eine schwere Niereninsuffizienz entwickeln, sollte individuell beurteilt werden.
Hautreaktionen
Während der Anwendung von OSSEOR wurde über lebensbedrohliche Hautreaktionen (Stevens-Johnson-Syndrom (SJS), toxische epidermale Nekrolyse (NET) und Arzneimittelausschlag mit Eosinophilie und systemischen Symptomen (DRESS)) berichtet.
Die Patienten sollten über die Anzeichen und Symptome aufgeklärt und engmaschig auf Hautreaktionen überwacht werden. Das größte Inzidenzrisiko für SJS oder NET besteht innerhalb der ersten Behandlungswochen und innerhalb von 3-6 Wochen für DRESS.
Wenn interstitielle Anzeichen und Symptome von SJS oder NET (z. B. fortschreitender Hautausschlag, häufig mit Blasenbildung und Schleimhautläsionen) oder DRESS (z. B. Hautausschlag, Fieber, Eosinophilie und systemische Beteiligung (z. B. Adenopathie, Hepatitis, Nephropathie und Lungenerkrankung)) auftreten, OSSEOR sollte sofort gestoppt werden.
Die besten Ergebnisse bei der Behandlung von SJS, NET oder DRESS werden nach einer frühen Diagnose und sofortigem Absetzen jedes verdächtigen Arzneimittels erzielt. Ein frühzeitiger Abbruch der Behandlung ist mit einer besseren Prognose verbunden. Das klinische Bild von DRESS verschwand in den meisten Fällen nach Beendigung der Behandlung mit OSSEOR und gegebenenfalls Einleitung einer Kortikosteroidtherapie. Die Erholung kann langsam sein, und in einigen Fällen wurde nach Absetzen der Kortikosteroidtherapie über Rückfälle des Syndroms berichtet.
Bei Patienten, die unter Anwendung von OSSEOR SJS, NET oder DRESS entwickelt haben, darf die Therapie mit OSSEOR nicht mehr wieder aufgenommen werden.
Bei Patienten asiatischer Abstammung wurde über eine höhere, wenn auch immer noch seltene Inzidenz von Überempfindlichkeitsreaktionen einschließlich Hautausschlag, SJS oder NET berichtet.
Wechselwirkungen mit Labortests
Strontium stört kolorimetrische Verfahren zur Bestimmung der Calciumkonzentration im Blut und im Urin. Daher müssen in der klinischen Praxis Methoden der Atomemissionsspektrometrie mit induktiv gekoppeltem Plasma oder der Atomabsorptionsspektrometrie verwendet werden, um eine genaue Bestimmung der Calciumkonzentrationen im Blut und im Urin zu gewährleisten.
Hilfsstoff
OSSEOR enthält Aspartam, eine Phenylalaninquelle, die für Patienten mit Phenylketonurie gefährlich sein kann.
04.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Nahrungsmittel, Milch und ihre Derivate sowie kalziumhaltige Arzneimittel können die Bioverfügbarkeit von Strontiumranelat um ca. 60 - 70 % reduzieren. Daher sollte zwischen der Anwendung von OSSEOR und diesen Arzneimitteln ein Abstand von mindestens zwei Stunden liegen (siehe Abschnitte 4.2 und 5.2).
Da zweiwertige Kationen auf gastrointestinaler Ebene mit oralen Tetracyclinen (z. B. Doxycyclin) und Chinolon-Antibiotika (z. B. Ciprofloxacin) einen schlecht resorbierbaren Komplex bilden können, wird die gleichzeitige Anwendung von Strontiumranelat mit diesen Arzneimitteln nicht empfohlen. Als Vorsichtsmaßnahme sollte OSSEOR während der Behandlung mit oralen Tetracyclinen oder Chinolon-Antibiotika abgesetzt werden.
Eine klinische Studie in vivo zu Arzneimittelwechselwirkungen hat gezeigt, dass die Einnahme von Aluminium- und Magnesiumhydroxiden in den zwei Stunden vor oder gleichzeitig mit Strontiumranelat zu einer leichten Abnahme der Resorption von Strontiumranelat (20-25% Abnahme der AUC) führte, während die Resorption blieb praktisch unverändert, wenn das Antazida zwei Stunden nach der Einnahme von Strontiumranelat verabreicht wurde. Es ist daher vorzuziehen, Antazida mindestens zwei Stunden nach der Einnahme von OSSEOR einzunehmen. Da es jedoch empfohlen wird, OSSEOR vor dem Zubettgehen einzunehmen, wenn dieses Dosierungsschema nicht anwendbar ist , die gleichzeitige Anwendung bleibt akzeptabel.
Bei oraler Vitamin-D-Supplementierung wurden keine Wechselwirkungen beobachtet.
In klinischen Studien wurde weder eine klinische Wechselwirkungen noch ein signifikanter Anstieg des Strontiumspiegels im Blut nachgewiesen mit Arzneimitteln, die in der gegenwärtigen Praxis häufig gleichzeitig mit OSSEOR verschrieben werden, einschließlich: nichtsteroidale Antirheumatika (einschließlich Acetylsalicylsäure) , Anilide (wie Paracetamol), H2-Blocker und Protonenpumpenhemmer, Diuretika, Digoxin und Herzglykoside, organische Nitrate und andere Vasodilatatoren gegen Herzerkrankungen, Kalziumkanalblocker, Betablocker, ACE-Hemmer, Angiotensin-II-Antagonisten, selektive Beta-2- adrenerge Rezeptoragonisten, orale Antikoagulanzien, Thrombozytenaggregationshemmer, Statine, Fibrate und Benzodiazepin-Derivate.
04.6 Schwangerschaft und Stillzeit
Schwangerschaft
Daten zur Anwendung von Strontiumranelat bei schwangeren Frauen liegen nicht vor.Tierstudien haben bei hohen Dosen reversible Knocheneffekte bei den Nachkommen von Ratten und Kaninchen gezeigt, die während der Trächtigkeit behandelt wurden (siehe Abschnitt 5.3). die Behandlung sollte abgebrochen werden.
Fütterungszeit
Physikalisch-chemische Daten legen nahe, dass Strontiumranelat in die Muttermilch übergeht.OSSEOR sollte während der Stillzeit nicht angewendet werden.
Fruchtbarkeit
In Tierstudien wurden keine Auswirkungen auf die männliche und weibliche Fertilität beobachtet.
04.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Strontiumranelat hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
04.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
OSSEOR wurde in klinischen Studien mit etwa 8.000 Personen untersucht. Die Langzeitsicherheit wurde in Phase-III-Studien bei postmenopausalen Frauen mit Osteoporose untersucht, die bis zu 60 Monate lang mit Strontiumranelat 2 g/Tag (n = 3.352) oder Placebo (n = 3.317) behandelt wurden. Das Durchschnittsalter zum Zeitpunkt der Aufnahme betrug 75 Jahre und 23% der eingeschlossenen Patienten waren zwischen 80 und 100 Jahre alt.
In einer gepoolten Analyse randomisierter placebokontrollierter Studien bei postmenopausalen osteoporotischen Patienten waren die häufigsten Nebenwirkungen Übelkeit und Durchfall, die im Allgemeinen zu Beginn der Behandlung berichtet wurden, ohne nennenswerten Unterschied zwischen den Gruppen in den späteren Stadien. Der Abbruch der Therapie ist hauptsächlich auf Übelkeit zurückzuführen.
Es gab keinen Unterschied in der Art der Nebenwirkungen zwischen den Behandlungsgruppen, unabhängig davon, ob die Patienten zum Zeitpunkt der Aufnahme jünger oder älter als 80 Jahre waren.
Tabelle der Nebenwirkungen
Die folgenden Nebenwirkungen wurden während klinischer Studien und/oder während der Anwendung von Strontiumranelat nach Markteinführung berichtet. Die folgenden Nebenwirkungen sind nach folgender Konvention aufgeführt: sehr häufig (≥ 1/10), häufig (≥ 1/100,
§ Die Häufigkeit in klinischen Studien war in der Arzneimittelgruppe und in der Placebogruppe ähnlich.
* In asiatischen Ländern als selten gemeldet.
# Bei Nebenwirkungen, die in klinischen Studien nicht beobachtet wurden, beträgt die Obergrenze des 95-%-Konfidenzintervalls nicht mehr als 3 / X, wobei X die Gesamtstichprobengröße aus allen klinischen Studien und relevanten Studien darstellt.
a Muskel-Skelett-Fraktion > 3-fache Obergrenze des Normbereichs. In den meisten Fällen normalisierten sich diese Werte spontan ohne Therapieänderung.
Beschreibung ausgewählter Nebenwirkungen
Venöse Thromboembolie
In Phase-III-Studien betrug die jährliche Inzidenz von venösen Thromboembolien (VTE)-Ereignissen, die über 5 Jahre beobachtet wurden, etwa 0,7 % mit einem relativen Risiko von 1,4 (95 % KI = [1,0; 2, 0]) bei Patienten, die mit Strontiumranelat im Vergleich zu Placebo behandelt wurden (siehe Abschnitt 4.4).
Herzinfarkt
In einer gepoolten Analyse placebokontrollierter randomisierter klinischer Studien bei postmenopausalen osteoporotischen Patienten wurde bei Patienten, die mit Strontiumranelat behandelt wurden, im Vergleich zu Patienten, die Placebo erhielten, ein signifikanter Anstieg von Myokardinfarkten beobachtet (1,7 % im Vergleich zu 1,1 %) mit einem relativen Risiko von 1,6 (95%-KI = [1,07; 2,38]).
Meldung von vermuteten Nebenwirkungen
Die Meldung von vermuteten Nebenwirkungen, die nach der Zulassung des Arzneimittels auftreten, ist wichtig, da sie eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem zu melden.
04.9 Überdosierung
Symptome
In einer klinischen Studie, in der die wiederholte Gabe von 4 g Strontiumranelat täglich über mehr als 25 Tage bei gesunden postmenopausalen Frauen untersucht wurde, wurde eine gute Verträglichkeit festgestellt. Die einmalige Gabe von Dosen bis zu 11 g bei jungen gesunden männlichen Probanden verursachte keine besonderen Symptome.
Verwaltung
Bei der Beobachtung von Überdosierungsepisoden in klinischen Studien (bis zu 4 g/Tag über einen maximalen Zeitraum von 147 Tagen) wurden keine klinisch relevanten Wirkungen beobachtet.
Die Gabe von Milch oder Antazida kann sinnvoll sein, um die Aufnahme des Wirkstoffs zu reduzieren.Bei einer erheblichen Überdosierung kann die Möglichkeit in Betracht gezogen werden, Erbrechen auszulösen, um den nicht resorbierten Wirkstoff zu eliminieren.
05.0 PHARMAKOLOGISCHE EIGENSCHAFTEN
05.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Arzneimittel zur Behandlung von Knochenerkrankungen - Andere Arzneimittel, die die Knochenstruktur und -mineralisierung beeinflussen, ATC-Code: M05BX03
Wirkmechanismus
In vitro, Strontiumranelat:
- erhöht die Knochenbildung in Knochengewebekulturen sowie die Replikation von Osteoblasten-Vorläufern und die Kollagensynthese in Knochenzellkulturen;
- verringert die Knochenresorption durch Verringerung der Osteoklastendifferenzierung und ihrer Resorptionsaktivität.
Dies bestimmt eine Neuausrichtung des Knochenumsatzes zugunsten seiner Bildung.
Die Aktivität von Strontiumranelat wurde in mehreren experimentellen Studien nachgewiesen, insbesondere bei intakten Ratten erhöht Strontiumranelat die trabekuläre Knochenmasse, die Anzahl und Dicke der Trabekel, was zu einer Verbesserung der Knochenfestigkeit führt.
Strontium wird hauptsächlich an der kristallinen Oberfläche resorbiert und ersetzt sowohl bei Tieren als auch bei Menschen in Behandlung das Calcium im Apatitkristall in neu gebildeten Knochen nur in begrenztem Umfang. Strontiumranelat verändert die Eigenschaften des Knochenkristalls nicht. In Beckenkammknochenbiopsien, die nach einer Behandlung mit Strontiumranelat 2 g/Tag über bis zu 60 Monate in Phase-III-Studien entnommen wurden, wurden keine schädlichen Auswirkungen auf die Knochenqualität oder Mineralisierung beobachtet.
Die kombinierten Effekte der Verteilung von Strontium im Knochen (siehe Abschnitt 5.2) und die höhere Röntgenabsorption von Strontium im Vergleich zu Calcium führen zu einer Erhöhung des Wertes der Knochendichtemessung (BMD), gemessen durch Doppelstrahl-Photonenabsorptiometrie ( DXA) . Die verfügbaren Daten weisen darauf hin, dass diese Faktoren für etwa 50 % der beobachteten Veränderungen der BMD über eine 3-jährige Behandlung mit OSSEOR 2 g/Tag verantwortlich sind. Dies sollte bei der Beurteilung von Veränderungen der Knochendichte während der Behandlung mit OSSEOR berücksichtigt werden. In den Phase-III-Studien, in denen die Wirksamkeit der Behandlung mit OSSEOR bei der Reduzierung von Frakturen nachgewiesen wurde, erhöhte OSSEOR die mittlere Knochendichte ab Einschluss um ca. 4 % jährlich an den Lendenwirbeln und um 2 % jährlich an den Lendenwirbeln des Oberschenkelhalses, die , je nach Studie 13 bis 15 % bzw. 5 bis 6 % nach 3 Jahren.
In Phase-III-Studien waren im Vergleich zu Placebo die biochemischen Marker der Knochenbildung (spezifische alkalische Phosphatase und C-terminales Propeptid von Typ-I-Prokollagen) und die der Knochenresorption (Serum-C-Telopeptid und Urin-Crosslinks von N-Telopeptid) erhöht. vom dritten Monat bis zum dritten Behandlungsjahr zurückgegangen.
Zusätzlich zu den primären pharmakologischen Wirkungen von Strontiumranelat wurden eine leichte Abnahme der Serumcalcium- und Parathormonspiegel (PTH), eine Zunahme der Phosphorkonzentration im Blut und der gesamten Aktivität der alkalischen Phosphatase ohne klinische Konsequenzen beobachtet.
Klinische Wirksamkeit
Osteoporose ist definiert als BMD der Wirbelsäule oder Hüfte, die 2,5 oder mehr Standardabweichungen unter dem Mittelwert in der jungen Normalbevölkerung liegt. Einige Risikofaktoren sind mit postmenopausaler Osteoporose verbunden, darunter niedrige Knochenmasse, niedrige Knochenmineraldichte, frühe Menopause, Rauchen und eine Familienanamnese von Osteoporose. Die klinische Folge der Osteoporose sind Frakturen, wobei das Risiko für Frakturen mit steigender Zahl der Risikofaktoren steigt.
Behandlung der postmenopausalen Osteoporose
Das Studienprogramm zur Bewertung der Frakturreduktion mit OSSEOR bestand aus zwei placebokontrollierten Phase-III-Studien: der SOTI-Studie und der TROPOS-Studie. An der SOTI-Studie nahmen 1.649 postmenopausale Frauen mit dokumentierter Osteoporose (niedrige lumbale BMD und vorherrschende Wirbelfrakturen) und einem Durchschnittsalter von 70 Jahren teil. An der TROPOS-Studie nahmen 5.091 postmenopausale Frauen mit Osteoporose (niedrige Knochendichte des Oberschenkelhalses und mindestens eine Fraktur bei mehr als der Hälfte der Patienten) und einem Durchschnittsalter von 77 Jahren teil. In die Studien SOTI und TROPOS nahmen zusammen 1.556 Patienten ein, die zum Zeitpunkt des Einschlusses über 80 Jahre alt waren (23,1 % der Studienpopulation).In beiden Studien wurden zusätzlich zur Therapie (2 g / Tag Strontium oder Placebo) die Patienten Einnahme von ausreichend Kalzium- und Vitamin-D-Ergänzungen.
In der SOTI-Studie reduzierte OSSEOR das relative Risiko neuer Wirbelfrakturen um 41 % über 3 Behandlungsjahre (Tabelle 1). Der Effekt war ab dem ersten Jahr signifikant und zeigte ähnliche Vorteile bei Frauen mit multiplen Frakturen bei der Aufnahme in die Studie. In Bezug auf klinische Wirbelfrakturen (definiert als Frakturen mit Rückenschmerzen und/oder einer Abnahme der Körpergröße um mindestens 1 cm) wurde das relative Risiko um 38 % reduziert von mindestens 1 cm im Vergleich zu Placebo. Die Bewertung der Lebensqualität anhand der spezifischen QUALIOST-Skala sowie die allgemeinen Scores zur Gesundheitswahrnehmung der allgemeinen SF-36-Skala weisen auf den Nutzen von OSSEOR im Vergleich zu Placebo hin.
Die Wirksamkeit von OSSEOR bei der Reduzierung des Risikos neuer Wirbelfrakturen wurde durch die TROPOS-Studie auch bei osteoporotischen Patienten ohne Fragilitätsfrakturen zum Zeitpunkt des Einschlusses bestätigt.
Eine gemeinsame Analyse der Studien SOTI und TROPOS zeigte, dass OSSEOR bei Patienten, die zum Zeitpunkt der Aufnahme über 80 Jahre alt waren, das relative Risiko neuer Wirbelfrakturen über 3 Behandlungsjahre um 32 % reduzierte (Inzidenz von 19 ; 1 % mit Strontiumranelat vs. 26,5% mit Placebo).
In einer Analyse in der Folge der Patienten in den SOTI- und TROPOS-Studien mit Lendenwirbel- und/oder Oberschenkelhals-BMD im osteopenischen Bereich zum Zeitpunkt des Einschlusses und ohne prävalente Frakturen, aber mit mindestens einem zusätzlichen Frakturrisikofaktor (N = 176), reduzierte OSSEOR das Risiko einer ersten Wirbelfraktur um 72 % über 3 Jahre (Inzidenz einer Wirbelfraktur 3,6 % unter Strontiumranelat vs. 12,0 % unter Placebo).
Eine Analyse in der Folge wurde in einer Untergruppe von TROPOS-Patienten von besonderem medizinischem Interesse und mit hohem Frakturrisiko [definiert als Patienten mit einem Oberschenkelhals-BMD-T-Score ≤-3 SD (Herstellerbereich entsprechend -2,4 SD nach NHANES III) und einem " Alter > 74 Jahre (n = 1.977, dh 40% der TROPOS-Studienpopulation)].
In dieser Gruppe reduzierte OSSEOR nach mehr als 3 Behandlungsjahren das Risiko von Hüftfrakturen um 36 % im Vergleich zu Placebo (Tabelle 2).
Behandlung von Osteoporose beim Menschen
Die Wirksamkeit von OSSEOR wurde bei Männern mit Osteoporose in einer doppelblinden, placebokontrollierten 2-Jahres-Studie nachgewiesen, wobei eine Hauptanalyse nach einem Jahr bei 243 Patienten (Population Absicht zu behandeln, 161 mit Strontiumranelat behandelte Patienten) mit hohem Frakturrisiko (mittleres Alter 72,7 Jahre; mittlere lumbale BMD mit einem T-Score von -2,6; 28% prävalente Wirbelfraktur).
Alle Patienten erhielten täglich Kalzium- (1000 mg) und Vitamin D (800 IE) Ergänzungen.
Statistisch signifikante Erhöhungen der BMD-Werte wurden bereits 6 Monate nach Behandlungsbeginn mit OSSEOR im Vergleich zu Placebo beobachtet.
Ein statistisch signifikanter Anstieg der mittleren BMD-Werte der Lendenwirbelsäule wurde über den 12-Monats-Zeitraum beobachtet, das Hauptwirksamkeitskriterium (E (SE) = 5,32 %; 95 %-KI = [3,86; 6,79]: p Menopause.
Statistisch signifikante Erhöhungen der BMD des Oberschenkelhalses und der Gesamt-BMD des Femurs wurden beobachtet (p
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat auf die Verpflichtung zur Vorlage von Studienergebnissen mit OSSEOR in allen Untergruppen der pädiatrischen Population bei Osteoporose verzichtet (siehe Abschnitt 4.2 für Informationen zur pädiatrischen Anwendung).
05.2 „Pharmakokinetische Eigenschaften
Strontiumranelat besteht aus 2 stabilen Strontiumatomen und einem Molekül Ranelsäure, einer organischen Komponente, die hinsichtlich Molekulargewicht, Pharmakokinetik und Akzeptanz des Arzneimittels den besten Kompromiss darstellt. Die Pharmakokinetik von Strontium und Ranelsäure wurde bei gesunden jungen männlichen Freiwilligen, bei gesunden postmenopausalen Frauen und während der Langzeitbehandlung bei Männern mit Osteoporose und bei Frauen mit postmenopausaler Osteoporose, einschließlich älterer Menschen, untersucht.
Die Aufnahme, Verteilung und Bindung von Ranelsäure an Plasmaproteine ist aufgrund ihrer hohen Polarität gering. Es gibt keine Akkumulation von Ranelsäure und keine Hinweise auf eine Metabolisierung bei Tieren und Menschen Resorbierte Ranelsäure wird unverändert schnell über den Urin ausgeschieden.
Absorption
Die absolute Bioverfügbarkeit von Strontium beträgt 25 % (Bereich 19-27 %) nach einer oralen Dosis von 2 g Strontiumranelat. Die maximalen Plasmakonzentrationen werden 3-5 Stunden nach einer Einzeldosis von 2 g erreicht.
Der Steady-State wird nach 2 Wochen Behandlung erreicht. Die Einnahme von Strontiumranelat zusammen mit Calcium oder Nahrung reduziert die Bioverfügbarkeit von Strontium um ca. 60 - 70 % im Vergleich zur Einnahme 3 Stunden nach einer Mahlzeit Aufgrund der relativ langsamen Resorption von Strontium sollte eine Nahrungsaufnahme und Calciumaufnahme vor und nach der Einnahme von OSSEOR vermieden werden . Eine orale Vitamin-D-Supplementierung beeinträchtigt die Strontium-Exposition nicht.
Verteilung
Strontium hat ein Verteilungsvolumen von ca. 1 l/kg. Die Bindung von Strontium an humane Plasmaproteine ist gering (25%) und Strontium hat eine „hohe Affinität zu Knochengewebe. Messung der Strontiumkonzentration in Beckenkammknochenbiopsien von Patienten, die bis zu 60 Monate mit 2 g/Tag Strontiumranelat behandelt wurden , zeigt, dass die Strontiumkonzentration im Knochen nach etwa 3 Behandlungsjahren ein Plateau erreichen kann. Es liegen keine Patientendaten vor, die die Kinetik der Elimination von Strontium aus dem Knochen nach Absetzen belegen.
Biotransformation
Als zweiwertiges Kation wird Strontium nicht metabolisiert. Strontiumranelat hemmt den Cytochrom-P450-Enzymkomplex nicht.
Beseitigung
Die Elimination von Strontium ist zeit- und dosisunabhängig Die effektive Halbwertszeit von Strontium beträgt ca. 60 Stunden. Die Ausscheidung von Strontium erfolgt über die Nieren und den Magen-Darm-Trakt, seine Plasmaclearance beträgt ca. 12 ml/min (VK 22%) und seine renale Clearance ca. 7 ml/min (VK 28%).
Pharmakokinetik in bestimmten Populationen
Ältere Patienten
Populationspharmakokinetische Daten zeigten keine Korrelation zwischen dem Alter und der scheinbaren Clearance von Strontium in der Zielpopulation.
Nierenversagen
Bei Patienten mit mittelschwerer bis mittelschwerer Nierenfunktionsstörung (Kreatinin-Clearance 30-70 ml/min) nimmt die Strontium-Clearance mit abnehmender Kreatinin-Clearance ab (ca. 30 % Abnahme im Bereich der Kreatinin-Clearance von 30 bis 70 ml/min); dies führt zu einer Erhöhung in den Plasma-Strontiumspiegeln.In Phase-III-Studien hatten 85 % der Patienten eine Kreatinin-Clearance zwischen 30 und 70 ml/min, 6 % weniger als 30 ml/min bis zum Einschluss und die mittlere Kreatinin-Clearance betrug 50 ml/min. Bei Patienten mit mittelschwerer bis mittelschwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich Es liegen keine pharmakokinetischen Daten bei Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance
Leberinsuffizienz
Bei Patienten mit Leberinsuffizienz liegen keine pharmakokinetischen Daten vor.Aufgrund der pharmakokinetischen Eigenschaften von Strontium ist keine Wirkung zu erwarten.
05.3 Präklinische Sicherheitsdaten
Präklinische Daten lassen auf der Grundlage konventioneller Studien zu keine besonderen Gefahren für den Menschen erkennen Sicherheitspharmakologie, Genotoxizität, kanzerogenes Potenzial.
Bei Nagetieren führte die chronische orale Verabreichung hoher Dosen von Strontiumranelat zu Knochen- und Zahnanomalien, die hauptsächlich aus spontanen Frakturen und verzögerter Mineralisation bestanden, die nach Absetzen der Behandlung reversibel waren. Diese Wirkungen wurden bei einem Strontiumspiegel in den Knochen gefunden, der 2-3 mal höher war als der beim Menschen nach einer Behandlung von bis zu 3 Jahren.
Entwicklungstoxizitätsstudien führten bei Nachkommen von Ratten und Kaninchen zu Knochen- und Zahnanomalien (z. B. Verbiegung langer Röhrenknochen und gewellter Rippen), die 8 Wochen nach Beendigung der Behandlung reversibel sind.
Umweltrisikobewertung (ERA)
Die Umweltrisikobewertung von Strontiumranelat wurde in Übereinstimmung mit den europäischen Richtlinien zum ERA durchgeführt.
Strontiumranelat stellt keine Gefahr für die Umwelt dar.
06.0 PHARMAZEUTISCHE INFORMATIONEN
06.1 Hilfsstoffe
Aspartam (E951)
Maltodextrin
Mannit (E421)
06.2 Inkompatibilität
Nicht relevant.
06.3 Gültigkeitsdauer
- 3 Jahre.
- Nach Rekonstitution in Wasser ist die Suspension 24 Stunden lang stabil. Es wird jedoch empfohlen, die Suspension unmittelbar nach der Zubereitung zu trinken (siehe Abschnitt 4.2).
06.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Lagerbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
06.5 Art der unmittelbaren Verpackung und Inhalt des Packstücks
Papier-/Polyethylen-/Aluminium-/Polyethylenbeutel.
Pakete
Packungen mit 7, 14, 28, 56, 84 oder 100 Beuteln.
Es werden möglicherweise nicht alle Packungsgrößen in den Verkehr gebracht.
06.6 Gebrauchs- und Handhabungshinweise
Keine besonderen Anweisungen.
07.0 INHABER DER MARKETING-ERLAUBNIS
LES LABORATOIRES-SERVIER
50, rue Carnot
92284 Suresnes cedex
Frankreich
08.0 NUMMER DER MARKETING-ERLAUBNIS
EU / 1/04/287/001
EU / 1/04/287/002
EU / 1/04/287/003
EU / 1/04/287/004
EU / 1/04/287/005
EU / 1/04/287/006
036588061
036588022
036588034
036588046
036588010
036588059
09.0 DATUM DER ERSTEN GENEHMIGUNG ODER ERNEUERUNG DER GENEHMIGUNG
Datum der Erstzulassung: 21. September 2004
Datum der letzten Verlängerung: 21. September 2014